
Газета «Новости медицины и фармации» Гастроэнтерология. Проктология (649) 2018 (тематический номер)
Вернуться к номеру
Молекулярно-генетическая диагностика синдрома Жильбера
Авторы: Мельник А.А. – к.б.н.
Рубрики: Гастроэнтерология
Разделы: Справочник специалиста
Версия для печати
Синдром Жильбера (СЖ) — распространенное генетически детерминированное наследственное заболевание, связанное с конъюгацией билирубина (разновидность непрямой гипербилирубинемии), симптомом которого является периодически проявляющаяся умеренная желтуха при воздействии неблагоприятных факторов внешней и внутренней среды (физическое напряжение, инфекции, психические стрессы, низкокалорийная диета). Впервые этот синдром был описан французским врачом A. Gilbert [1] в начале XX века. Частота встречаемости СЖ в европеоидной, негроидной и монголоидной расах составляет соответственно 5–10, 30–35 и 2–3 % [2, 3]. По статистическим данным, этот синдром чаще проявляется у мужчин (12,4 %) по сравнению с женщинами (4,8 %) с соотношением 3 : 1–4 : 1. Средняя концентрация билирубина при СЖ значительно выше у мужчин, что может быть связано с андрогенным ингибированием стероидами процесса билирубин-ферментативного глюкуронирования. СЖ передается по аутосомно-рецессивному типу наследования, и, как правило, чаще всего манифестация наблюдается в возрасте до 30 лет. Основным клиническим проявлением СЖ является желтушное окрашивание кожи, склер, слизистых. Во время эпизодов желтухи отмечаются боль в животе, судороги в желудке, вздутие живота, диарея или запор, чувство усталости, потеря аппетита, головокружение, депрессия, потливость. У пациентов возможна манифестация с развитием токсических реакций при приеме некоторых лекарственных препаратов: анаболических стероидов, глюкокортикоидов, андрогенов, рифампицина, циметидина, хлорамфеникола, стрептомицина, салицилата натрия, ампициллина, кофеина, этинилэстрадиола, парацетамола, иринотекана.
Источники билирубина
У здорового человека в сутки распадается около 1 % циркулирующих эритроцитов. Освободившийся при этом гемоглобин в клетках ретикулоэндотелиальной системы (костный мозг, селезенка) распадается на гем (небелковую железосодержащую часть, в молекуле которой железо связано с протопорфирином) и белковое соединение — глобин. Основная часть билирубина (80–85 %) синтезируется в печени, а около 15–20 % — из других источников, таких как гемопротеины (миоглобин, цитохромы, каталаза, пероксидаза), и из незрелых клеток (эритробласты, незрелые ретикулоциты). В организме человека ежедневно образуется 250–350 мг билирубина.
Ферментативный механизм образования билирубина
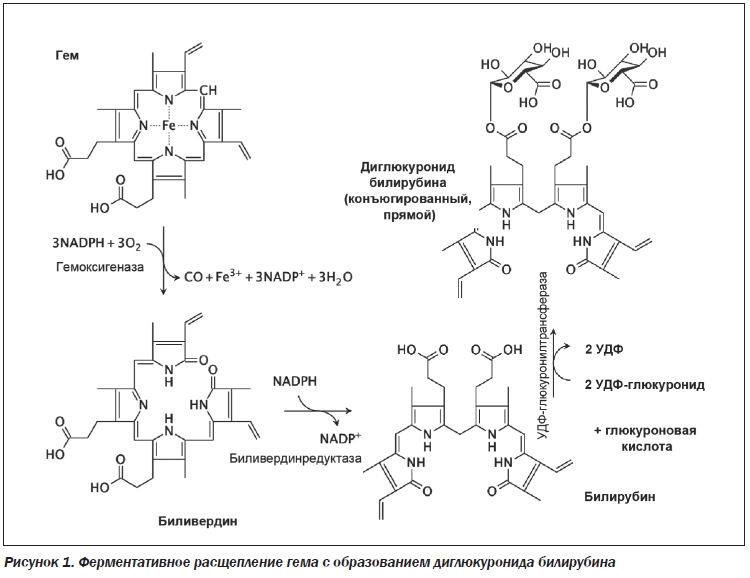
После распада гемоглобина в ретикулоэндотелиальной системе и образования гема и глобина происходит окисление гема под действием гемоксигеназы. В этой реакции расходуется три молекулы кислорода и НАДФ (никотинадениндинуклеотидфосфат) с образованием эквивалентного количества биливердина и оксида углерода. В дальнейшем биливердин под воздействием биливердинредуктазы конвертируется в билирубин. Образовавшийся билирубин (неконъюгированный, непрямой, свободный) является гидрофобным, липофильным соединением, не способным к почечной и билиарной секреции. Даже умеренно повышенный уровень свободного билирубина является токсичным для центральной нервной системы, что проявляется невыраженными клиническими симптомами, такими как ослабление внимания, утомляемость и др. Билирубин имеет высокое сродство к альбумину, поэтому 99 % неконъюгированного билирубина циркулирует в плазме в виде комплекса. Неконъюгированный билирубин, связанный с альбумином, транспортируется в печень, где происходит диссоциация этого комплекса. В гепатоцитах непрямой билирубин отщепляется от альбумина и взаимодействует с глюкуроновой кислотой при участии фермента уридиндифосфатглюкуронозилтрансферазы (UGT1A1), образуя при этом глюкуронид билирубина (моноглюкуронид — 15 % и диглюкуронид — 85 %) или конъюгированный (прямой) билирубин (рис. 1).
У большинства пациентов с СЖ гипербилирубинемия проявляется в молодом возрасте. В зависимости от референтных интервалов, установленных в лабораториях, верхний предел для общего билирубина в норме — от 17 до 20 мкмоль/л, из которых 75 % приходится на неконъюгированный билирубин [4]. При СЖ концентрация общего билирубина составляет от 20 до 50 мкмоль/л и редко превышает 85 мкмоль/л.
Несмотря на развитие новых медицинских технологий, диагностика СЖ до сих пор остается достаточно сложной. При наличии огромного количества лабораторных (неконъюгированный билирубин, общий анализ крови с ретикулоцитами, ферменты печени) и функциональных (гипокалорийный, с никотиновой кислотой, с фенобарбиталом) тестов, не выработан оптимальный алгоритм применения, основанный на их чувствительности и специфичности. В связи с этим рекомендуется проведение генетического анализа для подтверждения синдрома Жильбера.
Клиническая картина СЖ обусловлена снижением до 30 % уровня функциональной активности уридиндифосфатглюкуронозилтрансферазы — фермента, необходимого для конъюгирования билирубина. Этот фермент кодируется геном UGT1A, расположенным на 2q37 хромосоме.
Ген UGT1A
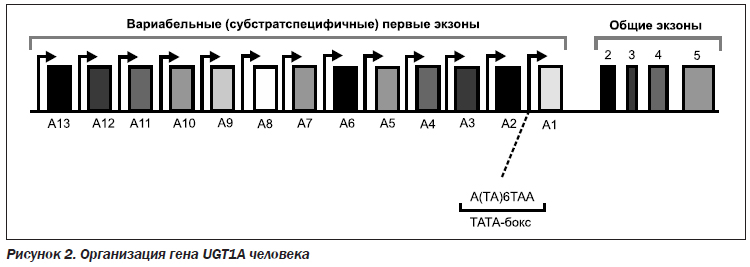
В 1991 г. J. Ritter и др. [5] сообщили о выделении из печени двух клонов, кодирующих ДНК, — UGT1A и UGT1D (переименованные в дальнейшем в UGT1A и UGT1A4) с билирубиновой глюкуронилтрансферазной активностью. Нуклеотидная последовательность из 30 регионов этих двух клонов была одинаковой (кодируются одним геном), что и было подтверждено спустя год, когда эта группа сообщила о выделении большого участка гена UGT1A с его уникальной структурой [6]. В начале 2000-х годов был полностью изучен принцип работы гена UGT1A [7]. Ген UGT1A имеет длину 218 kb и содержит 13 аминотерминальных экзонов, которые кодируют субстрат, связывающий домен и 4 общих карбокситерминальных экзона, кодирующих УДФ-глюкуроновую кислоту.
Вариабельные экзоны 1A2–1A13 не участвуют в метаболизме билирубина. Генетические мутации, связанные с отсутствуем или снижением ферментативной активности, которые вызывают недостаточность при конъюгировании билирубина, локализованы в вариабельных экзонах 1А1 и общих экзонах со 2-го по 5-й (рис. 2).
Уридиндифосфатглюкуронозил-трансфераза (UGT1A1)
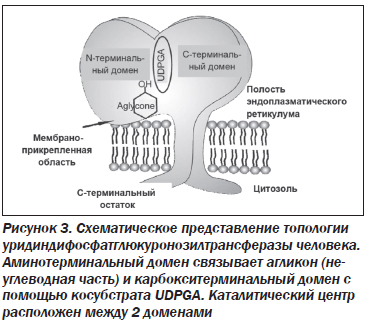
Ген UGT1A1 кодирует фермент уридиндифосфатглюкуронозилтрансферазу (UGT1A1). В печени этот фермент катализирует реакцию соединения (конъюгирования) билирубина (непрямой, неконъюгированный) с глюкуроновой кислотой. Данное состояние обусловлено мутацией в гене UGT1A. Активность UGT1A1 у пациентов с синдромом Жильбера составляла около 30 % от нормы, основанной на исследованиях с использованием образцов печени человека [8, 9]. В настоящее время семейство ферментов UGT1A1 продолжают активно изучать. Изоформы UGT1A обнаружены в различных отделах организма человека: в печени — UGT1А1, -1А3, -1А4, -1А6, -1А7, -1А9, в пищеводе и желудке — UGT1А8, в кишечнике — UGT1А10. Функцией ферментов семейства UGT1А1 является конъюгация как эндогенных метаболитов, гормонов, нейротрансмиттеров, так и экзогенных (различных ксенобиотиков, канцерогенов, лекарственных препаратов) [10, 11]. Семейство UGT человека (ЕС 2.4.1.17) имеет молекулярный вес ~56 кДа, в состав которых входит ~530 аминокислот. Они являются интегральными мембранными белками эндоплазматического ретикулума, состоящими из 2 аналогичных по размеру доменов (рис. 3).
Полиморфизм UGT1A1
У человека подсемейства UGT1 и UGT2 содержат 19 различных изоферментов, имеющих близкую последовательность [12, 13]. Однако только UGT1A1 способен глюкуронировать билирубин [14]. В 1995 г. было показано, что мутация в промоторном участке гена UGT1A1 является необходимой, но недостаточной для полной манифестации СЖ. Эта мутация, снижающая активность фермента UGT1A1, представляет собой вставку двух нуклеотидов в ТАТА-боксе промотора гена. В этом случае нормальная последовательность A(TA)6TAA трансформируется в последовательность A(TA)7TAA. Шесть повторов TA (тимин — аденин) в области промотора A(TA)6TAA соответствуют нормальной функциональной активности фермента UGT. При вставке седьмого ТА-повтора A(TA)7TAA ухудшается связывание с транскрипционным фактором, в результате чего уменьшается экспрессия гена, что ведет к снижению функциональной активности фермента, которая проявляется непрямой (неконъюгированной) гипербилирубинемией. При наличии вставки TA в гомозиготном состоянии наблюдается снижение активности фермента на 30 % и конъюгации билирубина в гепатоцитах на 80 % по отношению к норме. Тенденция к прогрессирующему снижению активности фермента также отмечается при увеличении TA-повторов (например, до 8). В случае увеличения числа ТА-повторов до 8 в одном из аллельных генов или в обоих СЖ также считается подтвержденным — А(ТА)7 ТАА/А(ТА)8ТАА и А(ТА)8 ТАА/А(ТА)8ТАА. Активность фермента UGT1A1 находится в обратной зависимости от числа повторов.
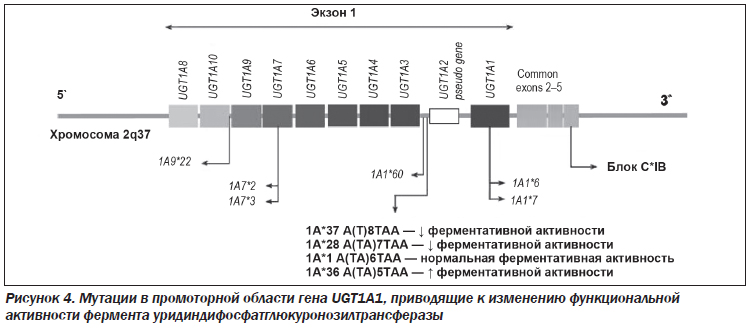
К настоящему времени для UGT1A1 показано существование не менее 4 аллелей: A(TA)6TAA (рис. 4):
1) UGT1A1*1 — «дикий тип» (wild type);
2) UGT1A1*28 — аллель A(TA)7TAA, наиболее частый мутантный;
3) UGT1A1*36 — аллель (TA)5;
4) UGT1A1*37 — аллель (TA)8.
Частота мутантного аллеля UGT1A1*28 в различных популяциях следующая: 26–39 % для европеоидов, 9–33 % для азиатской популяции, 40–56 % для негроидной популяции. Другие мутантные аллели — UGT1A1*36 и UGT1A1*37 — обнаружены главным образом в негроидной популяции с частотой 2–16 %.
Идентификация количества ТА-повторов в промоторной области гена UGT1A1
Самым быстрым и эффективным методом для выявления синдрома Жильбера является ДНК-диагностика с использованием метода ПЦР (полимеразная цепная реакция), заключающаяся в определении числа TA-повторов в гене UGT1A1.
1. Метод ПЦР с использованием капиллярного гель-электрофореза
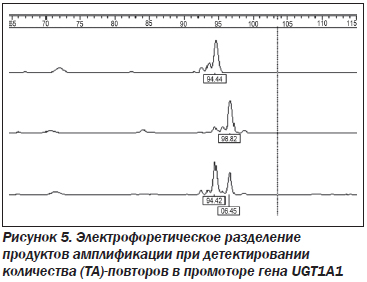
Молекулярно-генетическая диагностика проводится методом ПЦР с FAM-мечеными (карбоксифлуоресцеин) праймерами с разделением продуктов амплификации при помощи капиллярного гель-электрофореза.
Ампликон длиной 96 п.о. соответствует участку промотора гена UGT1A1 с 7 (ТА)-повторами, что позволяет подтвердить диагноз синдрома Жильбера. Ампликон длиной 94 п.о. соответствует нормальному участку промотора гена UGT1A1 с 6 (ТА)-повторами. Наличие двух ампликонов длиной 96 и 94 п.о. свидетельствует о гетерозиготном носительстве 6/7 (ТА)-повторов (рис. 5).
2. Секвенирование продуктов ПЦР
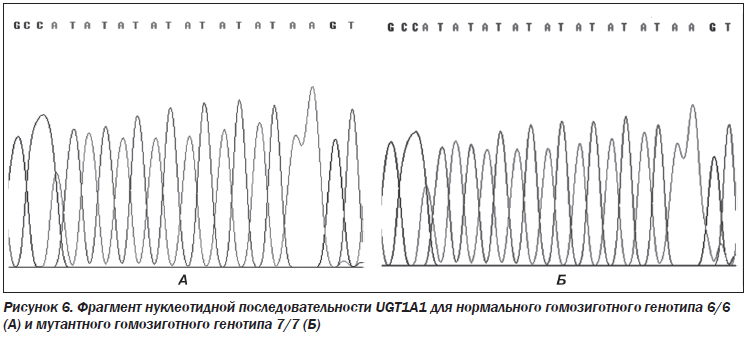
На рис. 6 представлены фрагмент нуклеотидной последовательности UGT1A1 для нормального гомозиготного генотипа 6/6 (структура повтора: A-(TA)6-TAA, результат секвенирования продукта ПЦР размером 97 п.н.) и фрагмент нуклеотидной последовательности UGT1A1 для мутантного гомозиготного генотипа 7/7 (структура повтора: A-(TA)7-TAA, результат секвенирования продукта ПЦР размером 99 п.н.).
Интерпретация результатов исследования
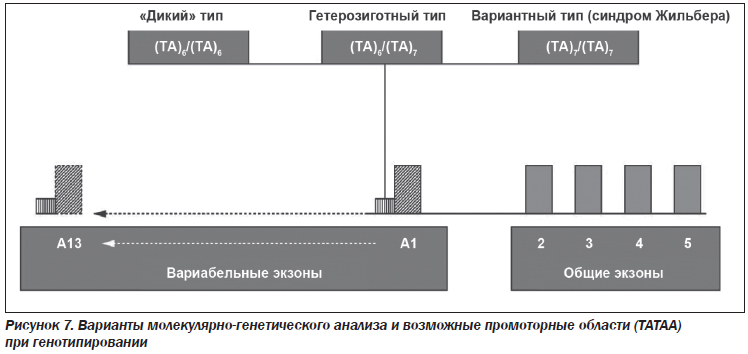
Варианты молекулярно-генетического анализа могут быть следующими (рис. 7):
1. UGT1A1 (ТА)6/(ТА)6 — нормальный («дикий») генотип. Синдром Жильбера не выявлен.
2. UGT1A1 (ТА)6/(ТА)7 — генотип с увеличением ТА-повторов в гене в гетерозиготном состоянии. Риск развития синдрома Жильбера в латентной или легкой форме.
3. UGT1A1 (ТА)7/(ТА)7 — генотип с увеличением ТА-повторов в гене в гомозиготном состоянии. Очень высокий риск развития синдрома Жильбера. Заболевание может протекать с более высоким повышением уровня билирубина и более тяжелыми клиническими проявлениями.
Заключение
В настоящее время все большее значение приобретает необходимость лабораторного подтверждения синдрома Жильбера, что связано с постановкой дифференциального диагноза. При выявлении повышенного уровня билирубина в сыворотке крови в течение продолжительного времени рекомендуется прежде всего выполнить пациенту молекулярно-генетический анализ. Назначение исследования ТА-повторов в гене UGT1A важно в следующих случаях: при дифференциальной диагностике СЖ от других заболеваний, сопровождающихся гипербилирубинемией, перед началом лечения с использованием лекарственных препаратов, обладающих гепатотоксическим действием, при возможности осложнений терапии иринотеканом, слабовыраженной неинфекционной желтухе, отягощенном семейном анамнезе и др. Необходимость назначения молекулярно-генетического анализа и интерпретации полученных результатов определяется врачом-генетиком при проведении медико-генетического консультирования.
В Украине анализ для определения ТА-повторов в промоторной области гена UGT1A1, ответственного за развитие синдрома Жильбера, можно сделать в некоторых частных медицинских лабораториях.
Список литературы
1. Gilbert A., Lereboullet P. La cholemie simple familiale // Semaine Medicale. — 1901. — 21. — 241-3.
2. Owens D., Evans J. Population studies on Gilbert’s syndrome // J. Med. Genet. — 1975. — 12. — 152-156.
3. Olsson R., Bliding A., Jagenburg R. et al. Gilbert’s syndrome — does it exist? A study of the prevalence of symptoms in Gilbert’s syndrome // Acta Med Scand. — 1988. — 224. — 485-90.
4. Dufour D.R., Lott J.A., Nolte F.S., Gretch D.R., Koff R.S., Seeff L.B. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests // Clin. Chem. — 2000. — 46. — 2027-49.
5. Ritter J.K., Crawford J.M., Owens I.S. Cloning of two human liver bilirubin UDP-glucuronosyltransferase cDNAs with expression in COS-1 cells // J. Biol. Chem. — 1991. — 266. — 1043-1047.
6. Ritter J.K., Chen F., Sheen Y.Y., Tran H.M., Kimura S., Yeatman M.T. et al. A novel complex locus UGT1 encodes human bilirubin phenol and other UDP-glucuronosyltransferase isozymes with identical carboxyl termini // J. Biol. Chem. — 1992. — 267. — 3257-3261.
7. Gong Q.H., Cho J.W., Huang T., Potter C., Gholami N., Basu N.K. et al. Thirteen UDP glucuronosyltransferase genes are encoded at the human UGT1 gene complex locus // Pharmacogenetics. — 2001. — 11. — 357-368.
8. Adachi Y., Yamamoto T. Hepatic bilirubin-conjugating enzymes of man in the normal state and in liver disease // Gastroenterol. Jpn. — 1982. — 17. — 235-240.
9. Raijmakers M.T., Jansen P.L., Steegers E.A., Peters W.H. Association of human liver bilirubin UDP-glucuronyltransferase activity with a polymorphism in the promoter region of the UGT1A1 gene // J. Hepatol. — 2000. — 33. — 348-351.
10. Strassburg C.P., Nguyen N., Mannss M.P., Tukey R.H. UDP-glucuronosyltransferase activity in human liver and colon // Gastroenterology. — 1999. — 116. — 149-60.
11. Strassburg C.P. Gilbert-Meulengracht’s syndrome and pharmacogenetics: is jaundice just the tip of the iceberg? // Drug Metabolism. Reviews. — 2010. — 42(1). — 168-81.
12. Mackenzie P.I., Bock K.W., Burchell B., Guillemette C., Ikushiro S., Iyanagi T., Miners J.O., Owens I.S., Nebert D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily // Pharmacogenet. Genomics. — 2005. — 15. — 677-685.
13. Canu G., Minucci A., Zuppi C., Capoluongo E. Gilbert and crigler najjar syndromes: An update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database // Blood Cells Mol. Dis. — 2013. — 50. — 273-280.
14. Bosma P.J., Seppen J., Goldhoorn B., Bakker C., Oude Elferink R.P., Chowdhury J.R., Chowdhury N.R., Jansen P.L. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man // J. Biol. Chem. — 1994. — 269. — 17960-17964.