Введение
Первичный латеральный склероз (ПЛС) представляет собой прогрессирующее нейродегенеративное заболевание верхних моторных нейронов, характеризующееся прогрессирующей спастичностью. Вовлекаются нижние конечности, туловище, верхние конечности и бульбарные мышцы (обычно в этом порядке).
Классификация болезней моторных нейронов
Термин «болезни двигательного нейрона» объединяет группу заболеваний с разными клиническими проявлениями и прогнозами, в основе которых лежит поражение двигательных нейронов головного и спинного мозга.
На сегодняшний день существует следующая классификация болезней мотонейронов (БМН), представленная A.J. Lerner в 2006 году:
1. Сочетанное вовлечение верхнего и нижнего мотонейронов:
— боковой амиотрофический склероз (БАС) (спорадический, семейный взрослых, семейный ювенильный).
2. Исключительное вовлечение нижнего мотонейрона:
— острая проксимальная наследственная моторная нейропатия (Верднига — Гоффмана);
— хроническая проксимальная наследственная моторная нейропатия у взрослых (Кугельберга — Веландера);
— наследственный бульбарный паралич (Х-сцеп-ленный, бульбоспинальная нейропатия Кеннеди);
— с глухотой (Фацио — Лонде);
— мультифокальная моторная нейропатия;
— постполиомиелитический синдром;
— пострадиационный синдром;
— фокальная спинальная мышечная атрофия.
3. Исключительное вовлечение верхнего двигательного нейрона:
— первичный латеральный склероз;
— наследственная спастическая параплегия.
БАС является наиболее распространенным из БМН. Согласно данным F. Norns (1993), в 80 % случаев болезнь двигательного нейрона представлена боковым амиотрофическим склерозом, в 10 % — прогрессирующим бульбарным параличом (ПБП), в 8 % — прогрессирующей мышечной атрофией (ПМА) и в 2 % — первичным латеральным склерозом. При этом средняя продолжительность жизни пациентов после начала заболевания при БАС составляет 3,3 года, при ПБП — 2,2 года. ПМА и ПЛС — более доброкачественные формы, текущие до 10 лет и более.
Что известно о первичном латеральном склерозе?
Это исключительно редкое заболевание.
Точные данные о частоте недостаточны: по разным источникам, от 0,5 до 4 % от болезней двигательных нейронов.
Триггерные факторы ПЛС, как и факторы риска, на сегодняшний день точно не установлены.
Семейный анамнез отсутствует, это исключительно спорадические случаи.
Развивается в любом возрасте, но обычно возникает в возрасте от 40 до 60 лет (возраст риска — 45 лет).
Один из вариантов первичного латерального склероза — это ювенильный первичный латеральный склероз. Начинается в раннем детстве и вызывается унаследованным патологическим геном от родителей: мутация в гене ALS2 (2q33-q35), что кодирует алсина (белок содержится в двигательных нейронах), иногда может возникать мутация в гене ERLIN2 (8p11.2).
Дисфункция и инвалидность нарастают по мере прогрессирования ПЛС. Медленная скорость прогрессирования ПЛС позволяет большинству пациентов и их семьям со временем адаптироваться к изменениям.
История понятия «первичный латеральный склероз»
По данным различных источников, впервые первичный латеральный склероз как самостоятельное заболевание был описан Шарко в 1865 г. В 1875 г. Эрб впервые предложил сам термин «первичный латеральный склероз». В 1904 г. Шпиллер сообщил о первичном латеральном склерозе как о синдроме медленно прогрессирующего восходящего спастического парапареза без признаков поражения периферического мотонейрона. В 1992 г. опубликованы критерии первичного латерального склероза (табл. 1).
Каково течение заболевания и на что важно обратить внимание?
Человек может часто терять равновесие и падать, так как при ПЛС в первую очередь нарушается походка. Иногда больные отмечают боль в шее, позвоночнике и ногах.
Повседневные дела, такие как уборка, переодевание, письмо и приготовление пищи, становятся более проблематичными из-за снижения подвижности.
Возникают проблемы с речью: она становится менее выразительной. Это связано с тем, что из-за слабости мышц глотки больному трудно контролировать мышцы языка, губ и гортани.
Из-за проблем с глотанием процесс приема пищи тоже усложняется.
В некоторых случаях наблюдается потеря эмоционального контроля с непредсказуемым смехом или плачем.
Клинический случай
Представляем клинический случай первичного латерального склероза. Больная Б., 61 год. Поступила в клинику Областного клинического центра неврологии и нейрохирургии г. Ужгорода 13.06.2017 в плановом порядке с жалобами на слабость в ногах и руках, больше слева, онемение левого бедра, головокружение, шаткость при ходьбе, частые падения, нарушение глотания, изменения голоса.
Анамнез заболевания. Считает себя больной около 3 лет, когда стала отмечать нарушения ощущений в ногах, появилась слабость в левой ноге, затем постепенно присоединилась слабость в левой руке и правых конечностях, в связи с чем перенесла несколько повторных падений. Обращалась в поликлинику центральной районной больницы по месту жительства, получала терапию по поводу остеохондроза пояснично-крестцового отдела позвоночника, без эффекта. За 6 месяцев до госпитализации отмечает нарастание слабости в нижних конечностях, стало трудно двигаться, появилось ощущение стягивания и тяжести в ногах. Обследована, в течение 8 месяцев неоднократно выполнялась магнитно-резонансная томография (МРТ) головного мозга — МР-признаки дисциркуляторной энцефалопатии; МРТ всех отделов позвоночника — дегенеративно-дистрофические изменения; ультразвуковое исследование брюшной полости — в пределах возрастной нормы. Трижды за 2016–2017 гг. находилась на стационарном лечении в условиях неврологического отделения Закарпатской областной клинической больницы им. А. Новака, где больной был выставлен диагноз «ишемическая миелопатия с нижним спастическим парапарезом», получала соответствующую терапию (миорелаксанты, сосудистая терапия, глюкокортикостероиды, витаминотерапия), без улучшения. На протяжении последних 2–3 недель до поступления в клинику Областного клинического центра нейрохирургии и неврологии (ОКЦНН) отмечает ухудшение состояния в виде расстройств глотания (поперхивается жидкостью), ухудшение зрения.
Анамнез жизни: не отягощен.
Неврологический статус на момент поступления. Сознание по шкале комы Глазго — 15 баллов. Менингеальные знаки отсутствуют. Контактна. MMSE — 26 баллов. Обращенную речь понимает, инструкции выполняет. Речь сохранена, с гнусавым оттенком. Глазные щели D = S, зрачки D = S. Движения глазных яблок: слабость конвергенции. Диплопия не отмечается, легкий горизонтальный нистагм. Лицо асимметрично, язык влево. Глотание нарушено: поперхивается жидкостью (проба трех ложек положительная). Глоточный рефлекс снижен. Тонус в нижних конечностях повышен по спастическому типу. Мышечная сила в конечностях снижена: в левой руке — 3 балла, в левой ноге — 3 балла, в правой руке — 3,5 балла, в правой ноге — 3 балла. Сухожильные рефлексы с рук высокие, D < S, с ног высокие с поликинетичным ответом, D ≤ S, клонусы стоп. Патологические кистевые (Россолимо, Якобсона — Ласка) и стопные (Бабинского) знаки с двух сторон. Мышечно-суставное чувство и вибрационная чувствительность сохранены. Брюшные рефлексы сохранены. Расстройств поверхностной чувствительности не показывает. В позе Ромберга практически не стоит. Тазовые функции не нарушены. Походка паретико-атактическая, с дополнительной опорой.
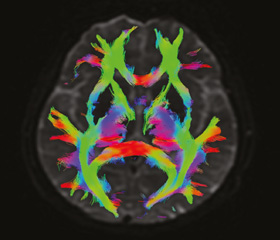
Дополнительные обследования в условиях клиники ОКЦНН. Общеклинические анализы: скорость оседания эритроцитов — 21 мм/ч. Реакция Вассермана — отрицательная. IgG к Borrelia burg. — не обнаружено. Витамин В12 — 525. Общий анализ ликвора: норма. Ликвор и кровь к HIV. Полимеразная цепная реакция ликвора: вирус герпеса 1/2 типа, Toxoplasma gondii, цитомегаловирус, вирус Эпштейна — Барр, вирус герпеса 6-го типа — не обнаружено. Аквапорин-4, антитела (АТ) IgG — отрицательно. Онкомаркеры (женская панель) — не выявлено. Креатинкиназа сыворотки крови — 606,8 Ед/л. МРТ шейно-грудного и поясничного отделов позвоночника: дегенеративно-дистрофические изменения позвоночника. МРТ головного мозга (рис. 1, 2): церебральная микроангиопатия, Fazekas score I. Электронейромиография: на момент обследования четких электромиографических данных генерализованного вовлечения переднего мотонейрона не получено. МР-трактография (рис. 2): снижение показателя FA кортикоспинального тракта на уровне PLIC мозга справа — 0,45, слева — 0,47, а также разрежение волокон кортикоспинального тракта с обеих сторон на уровне PLIC (значения FА кортикоспинальных трактов являются независимыми предикторами выживаемости).
/13-1.jpg)
Консультирована гинекологом: возрастная норма; окулистом: гипертоническая ангиопатия сосудов сетчатки; кардиологом: гипертоническая болезнь II; оториноларингологом: бульбарный синдром.
В отделении получала терапию: баклофен 40 мг в сутки; церебролизин 30 мл на 200 физр-ра внутривенно капельно, витаминотерапия, массаж. При выписке в неврологическом статусе без динамики.
Обсуждение
Дифференциальная диагностика проводилась с рядом заболеваний с учетом прежде всего признаков поражения мотонейронов, которые характеризуются развитием аналогичных симптомов в различных комбинациях: ПЛС, БАС, наследственная спастическая параплегия, оптикомиелит Девика, болезнь Марбурга, Лебера, поздняя форма амиотрофии Олбрайта.
Наследственная спастическая параплегия — наследственно обусловленная патология с поражением преимущественно пирамидных путей. Основным клиническим проявлением является прогрессирующий нижний спастический парапарез, при котором явления спастичности превалируют над выраженностью двигательных нарушений.
Оптикомиелит Девика — идиопатическое тяжелое воспалительное демиелинизирующее заболевание, характеризующееся избирательным вовлечением в патологический процесс зрительных нервов и спинного мозга при относительной интактности головного мозга. Первоначально в клинике наступают нарушения зрения в виде его снижения, вплоть до полной потери, через некоторое время присоединяются симптомы тяжелого поперечного миелита — парапарезы, тетрапарез, нарушение функции тазовых органов. Новыми критериями являются аутоантитела (аквапорин-4, АТ IgG).
Болезнь Марбурга (злокачественный рассеянный склероз, фульминантный рассеянный склероз) — поражает в основном лиц молодого возраста, характеризуется внезапным, острым началом, быстро прогрессирующим течением и отсутствием ремиссий. Неврологические нарушения отличаются преимущественным поражением ствола мозга с выраженными двигательными расстройствами (тетра-, гемиплегия), бульбарным синдромом (дисфония, дисфагия, дизартрия), глазодвигательными нарушениями (диплопия, парез взора вверх), с горметонией и децеребрационной ригидностью; кроме того, отмечают быстрое снижение когнитивных функций, также страдает острота зрения, развиваются различные виды афазии. Заболевание заканчивается летально в течение 1 года от начала первых симптомов. Диагностируется путем проведения МРТ головного мозга: выявляются псевдотуморозные очаги.
Болезнь Лебера (наследственная оптическая нейропатия Лебера, атрофия зрительных нервов Лебера) — наследственное заболевание, характеризующееся быстро или постепенно развивающимися двусторонними нарушениями центрального зрения у соматически здоровых молодых людей; наследуется по рецессивному типу. Является одним из признаков ряда наследственных патологических состояний, которые обусловлены отклонениями нормального функционирования дыхательной цепи митохондрий.
Поздняя форма амиотрофии Олбрайта. Спинальная мышечная атрофия — группа наследственных заболеваний, протекающих с поражением и потерей моторных нейронов передних рогов спинного мозга. Взрослые формы амиотрофий чаще всего развиваются в возрасте 15–60 лет, в среднем 35 лет, и имеют чаще всего доброкачественный характер. Согласно рекомендации Европейского консорциума по изучению нервно-мышечных заболеваний, клиническими критериями спинальной мышечной амиотрофии являются: симметричная мышечная гипотония и гипотрофия, фасцикуляции различных мышечных групп, гипо- или арефлексия мышц конечностей, отсутствие чувствительных, мозжечковых и интеллектуальных расстройств. Патогномоничных изменений при спинальной мышечной амиотрофии нет. Однако важно определение активности креатинкиназы сыворотки крови. При электронейромиографии выявляются симптомы поражения периферических моторных нейронов: спонтанная мышечная активность, увеличение длительности и амплитуды потенциалов действия двигательных единиц при нормальной скорости проведения импульсов по афферентным и эфферентным волокнам периферических нервов.
Боковой амиотрофический склероз (болезнь Шарко, болезнь Луи Герига) — хроническое прогрессирующее заболевание, при котором основными клиническими синдромами являются спастико-атрофические парезы конечностей и бульбарные расстройства, обусловленные сочетанным поражением центрального и периферического мотонейрона.
Диагностические критерии БАС (El Escorial)
Клинически достоверный БАС: признаки поражения верхнего (ВДН) и нижнего (НДН) двигательного нейрона в трех анатомических областях. Клинически достоверный БАС, лабораторно подтвержденный: признаки поражения ВДН и/или НДН в одной анатомической области, при этом пациент является носителем мутации дефектного гена.
Клинически вероятный БАС: признаки поражения ВДН и НДН в двух анатомических участках, с некоторыми симптомами привлечения ВДН, которые локализуются выше признаков вовлечения НДН. Клинически вероятный БАС, лабораторно подтвержденный: признаки поражения ВДН в одной и более анатомических областях и симптомы вовлечения НДН по данным электромиографии как минимум в двух анатомических областях.
Клинически возможный БАС: признаки поражения ВДН и НДН в одной анатомической области или признаки поражения ВДН как минимум в двух анатомических областях, или признаки поражения ВДН и НДН в двух анатомических областях без симптомов вовлечения ВДН выше признаков поражения НДН.
Дебют заболевания у нашей пациентки в возрасте до 60 лет в виде прогрессирующего тетрапареза, который начинался со слабости в нижних конечностях, с постепенным развитием слабости в верхних конечностях, нарастанием спастичности (ощущение стягивания и тяжести в ногах), постепенным присоединением бульбарных расстройств, клинически напоминает развитие и течение БАС, но отсутствие клинических проявлений поражения нижнего мотонейрона дало нам возможность исключить БАС. Кроме того, отсутствие наследственного анамнеза и клиническая картина поражения центрального мотонейрона позволили нам исключить и наследственный спастический парапарез. Отсутствие поражения зрительных нервов, характерных изменений (картины атрофии спинного мозга по всему его длиннику, особенно в каудальных отделах) при нейровизуализации и отрицательный анализ на аквапорин-4 окончательно позволили исключить диагноз «оптикомиелит Девика». Болезнь Лебера также была исключена, учитывая отсутствие наследственного фактора, течение заболевания, а также отсутствие характерных нарушений центрального зрения. Поздняя форма амиотрофии Олбрайта исключена на основании данных электронейромиографии и показателя креатинкиназы. В том числе исключили болезнь Марбурга, принимая во внимание возраст пациентки, клиническую картину, течение заболевания и, самое главное, данные МР-картины головного мозга.
Выводы
Таким образом, с учетом предыдущих попыток диагностического поиска данные дополнительных обследований, проведенных в условиях нашей клиники, позволили нам диагностировать у пациентки первичный латеральный склероз, с легким бульбарным синдромом, умеренно выраженным тетрапарезом, больше в левых конечностях, выраженными расстройствами походки и самообслуживания. Но на этом диагностический поиск не завершен.
БАС может первоначально дебютировать с наличием признаков поражения только верхнего или только нижнего мотонейрона. Таким образом, процесс, который изначально считается прогрессирующей мышечной атрофией или первичным латеральным склерозом, может быть переклассифицирован как БАС, если с течением времени развиваются достаточные признаки сочетания участия верхних и нижних моторных нейронов. В некоторых случаях такая переклассификация может происходить только при аутопсии.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов при подготовке данной статьи.

/12-1.jpg)