
Международный эндокринологический журнал 8 (64) 2014
Вернуться к номеру
Рекомендації Української асоціації кардіологів, Української асоціації ендокринних хірургів, Асоціації нефрологів України з диференційної діагностики артеріальних гіпертензій 2014 р.
Рубрики: Эндокринология
Разделы: Новости
Версия для печати
Статья опубликована на с. 125-154
Рекомендації підготувала робоча група з артеріальної гіпертензії Української асоціації кардіологів за участю спеціалістів–нефрологів та ендокринологів:
Проф. Свіщенко Є.П. (модератор, Київ)
Проф. Багрій А.Е. (Донецьк)
Проф. Єна Л.М. (Київ)
Проф. Коваль С.М. (Харків)
Академік НАМНУ Коваленко В.В. (Київ)
Д.м.н. Мелліна І.М. (Київ)
Проф. Сіренко Ю.М. (Київ)
За участю:
Член–кор. НАМНУ Колесник М.О. (Київ)
Проф. Дудар І.О. (Київ)
Проф. Ковальова О.М. (Харків)
Проф. Ларін О.С. (Київ)
Проф. Паньків В.І. (Київ)
Проф. Черенько С.М. (Київ)
Д.м.н. Радченко А.Д. (Київ)
К.м.н. Міщенко Л.А. (Київ)
К.м.н. Рековець О.Л. (Київ)
К.м.н. Товкай О.А. (Київ)
Зміст
Вступ
1. Ураження нирок та ниркових артерій, що можуть супроводжуватися розвитком АГ
1.1. Гломерулонефрит
1.2. Діабетична нефропатія
1.3. Хронічний пієлонефрит
1.4. Полікістоз нирок
1.5. Гідронефроз, обструктивна нефропатія
1.6. Гіпоплазія нирки
1.7. Ренінпродукуючі пухлини нирок
1.8. Травма нирки
1.9. Синдром Ліддла
1.10. Синдром Гордона
1.11. Реноваскулярна АГ
1.12. Довідкові матеріали
2. АГ, зумовлена ендокринними захворюваннями
2.1. Первинний альдостеронізм
2.2. Феохромоцитома
2.3. Синдром Кушинга
2.4. Акромегалія
2.5. Тиреотоксикоз
2.6. Гіпотиреоз
2.7. Гіперпаратиреоз
2.8. Синдром полікістозних яєчників
3. Гемодинамічні АГ
3.1. Коарктація аорти
3.2. Недостатність аортального клапана
4. Системні васкуліти
4.1. Синдром дуги аорти
4.2. Вузликовий поліартеріїт
5. АГ та синдром обструктивного апное сну
6. АГ, зумовлена неврологічними захворюваннями
6.1. Пухлини центральної нервової системи
6.2. Енцефаліт
6.3. Травми мозку
6.4. Спадкові порушення автономної регуляції
7. АГ, зумовлена медикаментами, наркотичними речовинами та харчовими компонентами
7.1. Кортикостероїди
7.2. Нестероїдні протизапальні засоби
7.3. Препарати жіночих та чоловічих статевих гормонів
7.4. Антидепресанти
7.5. Еритропоетин
7.6. Імуносупресанти
Вступ
Вторинна (симптоматична) артеріальна гіпертензія (АГ) є причиною підвищеного АТ у 5–10 % хворих на АГ. Ідентифіковано понад 50 захворювань та клінічних станів, що сприяють розвитку вторинної АГ. У більшості випадків така АГ характеризується високим АТ, несприятливим перебігом захворювання та високою частотою серцево–судинних катастроф. Проте при деяких її формах вчасна діагностика та адекватне лікування дозволяють досягти нормалізації АТ і запобігти розвитку ускладнень.
Поштовхом до видання даних Рекомендацій став аналіз статистичних даних МОЗ України щодо виявлення вторинних гіпертензій в нашій країні за період з 1999 по 2012 рік (рис. 1). Він наочно довів, що протягом останніх 14 років показник захворюваності на різні форми вторинної АГ суттєво знизився. Очевидно, що це зумовлено не зменшенням абсолютної кількості вторинних АГ, а погіршенням їх виявлення.
У наведених Рекомендаціях детально викладено діагностичні алгоритми та тактику лікування як найчастіших, так і рідкісних форм вторинних гіпертензій. Колектив авторів сподівається, що дані Рекомендації розширять уявлення лікарів про причини підвищення АТ і сприятимуть виявленню тих захворювань, при яких АГ є лише одним із симптомів, і які потребують специфічного лікування, результатом якого є нормалізація АТ.
1. Ураження нирок та ниркових артерій, що можуть супроводжуватися розвитком АГ
1.1. Гломерулонефрит (ГН)
Визначення та клінічна картина. Це група захворювань, які характеризуються двостороннім негнійним запаленням нирок, найчастіше імунної природи, що перебігають з ураженням клубочкового апарату та, як правило, з залученням до патологічного процесу інших ниркових структур. У сучасній нефрології термін «хронічний ГН» не використовується, це — збірне поняття, яке не може розглядатися як остаточний діагноз. Терміном «гломерулонефрит» визначаються такі гломерулярні ураження, як гострий постінфекційний ГН, швидко прогресуючий (підгострий та півмісяцевий) ГН, мезангіопроліферативний ГН (у т.ч. LgA–нефропатія та ін.). Стандартним методом діагностики ураження гломерулярного апарату є прижиттєве морфологічне дослідження ниркової тканини із встановленням гістологічної форми ГН.
Клінічна картина досить різноманітна і характеризується:
1) ізольованим сечовим синдромом (протеїнурія менше за 1 г/добу і мікрогематурія — частіше з гломерулярними зміненими еритроцитами в осаді сечі, можливо, циліндрами — гіаліновими, еритроцитарними, зернистими);
2) нефротичним синдромом (протеїнурія > 3 г/добу, гіпо– та диспротеїнемія, гіперліпідемія, набряки);
3) нефритичним синдромом/синдромом швидкопрогресуючого ГН, для якого характерна поява протеїнурії в межах сечового синдрому, еритроцитурії та циліндрурії різного ступеня вираженості, а також екстраренальні прояви: набряки та/або АГ; нерідко — порушення азотовидільної функції нирок, часто — швидкий розвиток і стрімке прогресування АГ та зниження функції нирок;
4) епізодами макрогематурії.
У частині випадків перебіг ГН латентний (практично без клінічних проявів), в інших — із загостреннями, для яких характерне поглиблення сечового синдрому, розвиток АГ або зростання її ступеня, зниження функції нирок або прогресування наявної ниркової недостатності.
Поширеність і характер АГ при ГН залежать від морфологічної та клінічної форми ГН і стадії ниркового ураження. АГ завжди має місце при наявності нефротичного синдрому (наприклад, у осіб із класичним варіантом гострого післяінфекційного ГН, при швидко прогресуючому ГН). При субклінічному перебігу гломерулярного процесу АГ може бути відсутня, її розвиток при ряді варіантів ГН нерідко за часом співпадає з етапом зниження функції нирок, однак у ряді випадків АГ може розвиватись і на ранній стадії ГН.
За різними даними, поширеність АГ при мезангіокапілярному ГН становить 35–80 %, при LgA–нефропатії — 43–74 %, при мембранозній гломерулопатії — 27–52 %, для нефропатії з мінімальними змінами АГ не характерна. АГ розглядається як важливий критерій несприятливого прогнозу: її наявність сприяє прискоренню темпів прогресування ниркового ураження та збільшенню ризику серцево–судинних ускладнень.
Вираженість АГ при ГН достатньо широко різниться. На етапах вираженого та тяжкого зниження функції нирок часто спостерігається резистентна АГ.
Механізми розвитку АГ багатофакторні; провідними серед них є:
1) активація ренін–ангіотензинової та симпатичної систем;
2) затримка натрію та води;
3) ремоделювання судин та ендотеліальна дисфункція;
4) на етапах зниження функції нирок — вторинний гіперпаратиреоз, анемія; в осіб на гемодіалізі — наявність артеріовенозного шунта.
Діагностика. Важливим при хронічній хворобі нирок, при ГН особливо, є ретельне динамічне лабораторне дослідження сечі. Оцінюються:
1) рівень та характер протеїнурії (інтермітуюча або персистуюча та ін.);
2) сечовий осад із визначенням наявності та ступеня вираженості лейкоцитурії (з екскрецією поліморфноядерних лейкоцитів або лімфоцитів), гематурії (що проявляється зміненими або незміненими гломерулярними еритроцитами або їх комбінуванням), циліндр–урії (гіалінові, зернисті, воскоподібні, еритроцитарні та ін.), ліпідурії та інших параметрів.
Необхідним є регулярний контроль рівня АТ (у т.ч. самоконтроль).
/126/126.jpg)
Інструментальні методи обстеження в діагностиці ГН є допоміжними. Вони використовуються переважно для виключення інших причин змін в аналізах сечі. Стандартним є регулярний контроль у динаміці розмірів нирок із використанням ультразвукового дослідження. Важливе значення в діагностиці та виявленні особливостей ГН, виборі лікувальної тактики та оцінці її ефективності надають прижиттєвому морфологічному дослідженню ниркової тканини (біопсії нирки) з використанням світлової, імунофлюоресцентної та електронної мікроскопії. Показаннями до проведення біопсії нирки є: 1) нефротичний синдром — практично в усіх дорослих хворих; 2) мікро– та макрогематурія — при наявності змінених еритроцитів (зазвичай > 50–80 %) та еритроцитарних циліндрів; а також ізольована мікрогематурія тривалістю понад 5 місяців, за умов відсутності вроджених анатомічних змін нирок або сечовивідних шляхів, сечокам’яної хвороби, кіст, пухлин нирок або сечовивідних шляхів; 3) гостре ураження нирок; 4) швидке прогресуюче зниження функції нирок у випадку невстановленого діагнозу, якщо розмір нирок ≥ 9 см; 5) системне захворювання сполучної тканини та васкуліти; 6) підозра на гостре або хронічне відторгнення ниркового трансплантата.
У процесі діагностики ГН необхідно враховувати головні клінічні характеристики тих системних захворювань сполучної тканини і системних васкулітів, перебіг яких супроводжується розвитком ГН:
1) системний червоний вовчак. Характерні: еритема на вилицях, дискоїдне висипання, фотосенсибілізація, виразки слизової оболонки ротової порожнини, ерозивний артрит ≥ 2 суглобів, серозит; різноманітна клінічна картина ГН — від латентного до швидкопрогресуючого; ураження центральної нервової системи — судоми або психоз; гематологічні (анемія, лейкопенія, тромбоцитопенія) та імунологічні (антитіла до нативної ДНК, антифосфоліпідні антитіла) порушення; підвищення рівня антинуклеарного фактора; критеріями діагнозу системного червоного вовчака є наявність 4 із вищеназваних критеріїв;
2) гранулематоз Вегенера (синусити та носові кровотечі; в легенях — інфільтрати, каверни, альвеолярні інфільтрати; ГН — часто по типу швидко прогресуючого; антитіла до антигенів цитоплазми нейтрофілів (ANCA);
3) мікроскопічний поліангіїт (пурпура, альвеолярні геморагії, полінейропатії);
4) синдром Чарга — Стросса (астма; еозинофілія; нейропатія; ГН — у частині випадків по типу швидко прогресуючого; ANCA).
Лікування. Терапія визначається морфологічною формою ГН, особливостями ниркового ураження та екстраренальних порушень, наявністю і ступенем АГ та ступенем зниження функції нирок. Лікувальна тактика при ГН визначається ренопротекторними та кардіопротекторними підходами, що включають:
— корекцію стилю життя: досягнення та утримання оптимальної ваги тіла, низькосольова дієта (< 5 г/добу кухонної солі, фізичні вправи (за відсутності протипоказань) 30 хв 5 разів на тиждень, інтенсивність яких сумісна з наявними у пацієнта серцево–судинними захворюваннями; відмова від паління;
— досягнення та підтримання цільового АТ < 140/90 мм рт.ст. для всіх хворих, крім хворих із протеїнурією, для яких цільовий САТ < 130 мм рт.ст., а також пацієнтів із цукровим діабетом — цільовий АТ < 140/85 мм рт.ст.;
— препаратами вибору для лікування АГ при ураженні нирок є інгібітори АПФ або блокатори АТ1–рецепторів ангіотензину ІІ (сартани), їх комбінації з діуретиками та блокаторами кальцієвих каналів, при необхідності застосовують інші класи медикаментозних засобів. Не допускається сумісне застосування інгібіторів АПФ і сартанів;
— відмова (або зведення до мінімуму) прийому нестероїдних протизапальних препаратів;
— контроль дисліпідемії статинами (цільовий рівень холестерину ліпопротеїнів низької щільності < 2,5 ммоль/л при високому або < 1,8 ммоль/л при дуже високому ризику серцево–судинних ускладнень);
— контроль гіперглікемії (цільовий рівень глікозильованого гемоглобіну — зазвичай менш 7,0 %);
— контроль анемії (препарати заліза, еритропоезстимулюючі засоби — до рівня гемоглобіну не більше 115 г/л);
— використання низькобілкової дієти;
— корекція порушень кальцієво–фосфорного балансу;
— за необхідності використовують різні режими імуносупресивної терапії (препарати цитотоксичної дії, глюкокортикоїди, можливо — протималярійні засоби);
— на етапах термінальної ниркової недостатності використовують лікувальні підходи, що заміняють функцію нирок (програмний гемодіаліз, перитонеальний діаліз, трансплантація нирки).
1.2. Діабетична нефропатія
Поширеність АГ в осіб із цукровим діабетом 1–го та 2–го типу з діабетичною нефропатією становить від 70 до 100 %. Діабетична нефропатія — класичне мікросудинне ускладнення цукрового діабету 1–го та 2–го типу з переважним ураженням гломерулярного апарату та тубулоінтерстиційними порушеннями (морфологічна картина нирок при діабетичній нефропатії в осіб із діабетом 1–го та 2–го типу має суттєві відмінності). Клінічно діабетична нефропатія характеризується протеїнурією, прогресуючим зниженням швидкості клубочкової фільтрації (ШКФ), особливо при значній протеїнурії, розвитком та прогресуванням АГ.
Механізми розвитку АГ. Окрім описаних у попередньому розділі механізмів, суттєвого значення у розвитку АГ при діабетичній нефропатії надають гіперглікемії.
Діагностика. Усім хворим на цукровий діабет рекомендується ретельний контроль та самоконтроль АТ. В осіб із цукровим діабетом 1–го та 2–го типу при відсутності анамнестичних та клінічних ознак ураження нирок обов’язковим є регулярний (не рідше 1 разу на рік) контроль загального аналізу сечі. При відсутності змін у таких аналізах — щорічне визначення мікроальбумінурії. Необхідним є регулярний контроль рівня креатиніну крові з розрахунком швидкості клубочкової фільтрації. Доцільним є ультразвукове дослідження нирок (розміри, структура) в динаміці. Прижиттєве морфологічне обстеження нирок (біопсія) проводиться рідше, ніж у хворих із ГН, переважно з метою виключення інших, не пов’язаних із діабетом, варіантів ниркового ураження (наприклад, швидкопрогресуючого та півмісяцевого ГН).
Лікування. Основою лікувальної тактики є застосування стандартних ренопротекторних підходів, що наведені у розділі, присвяченому лікуванню ГН. Особлива роль відводиться корекції гіперглікемії з досягненням цільового рівня глікозильованого гемоглобіну.
1.3. Хронічний пієлонефрит
Визначення та клінічна картина. Хронічний пієлонефрит — одно– або двостороннє неспецифічне інфекційно–запальне хронічне захворювання нирок, при якому в патологічний процес залучені ниркова чашечка (миска), чашечки та паренхіма нирки (в першу чергу і переважно — інтерстицій та канальцевий апарат), а також клубочки та ниркові судини. Хронічний пієлонефрит є причиною розвитку термінальної ниркової недостатності у 13–22 % випадків. Клінічна картина включає фази ремісії (при нормальному АТ і збереженій функції нирок може мати безсимптомний перебіг, також можуть бути відсутні зміни в аналізах сечі) та загострення (спостерігаються болі в поперековій ділянці та/або флангах живота, дизурія, часте сечовиділення, загальна слабкість, субфебрилітет; при тяжкому загостренні — гектична лихоманка, інтоксикаційний синдром). При хронічному пієлонефриті можуть спостерігатися симптоми, пов’язані з тубулоінтерстиціальними ураженнями (поліурія, ніктурія, збільшення екскреції натрію, зниження концентраційної функції нирок, гіперкаліємія, ацидоз). В аналізах сечі в період загострення зазвичай виявляється лейкоцитурія, гематурія (частіше — мікрогематурія), можуть виявлятися лейкоцитарні та бактеріальні циліндри. Протеїнурія, як правило, менша за 1 г/добу (при розвитку патологічних змін у гломерулярному апараті, зокрема, у вигляді фокального та сегментарного гломерулярного гіалінозу та склерозу можлива виражена протеїнурія в межах 1–3 г/добу і більше).
Механізми розвитку АГ: 1) підвищення активності ренін–ангіотензинової системи (частіше — при односторонньому ураженні); 2) гіперволемія (зазвичай — при двосторонніх процесах); 3) оклюзуюче ураження різних відділів судинного русла нирок (інтралобулярних та аркуантних артерій, потовщення стінки та фіброз інтими артеріол).
Поширеність АГ. АГ має місце у 50–75 % хворих із хронічним пієлонефритом, може розвиватись як при односторонньому, так і при двосторонньому ураженні нирок. Поширеність АГ більш висока в осіб зі зниженою функцією нирок. Для гострого пієлонефриту розвиток АГ не є характерним.
Діагностика. Хворі на хронічний пієлонефрит навіть за відсутності АГ потребують регулярного контролю АТ та періодичного (1–2 рази на рік при стабільному клінічному стані) контролю креатиніну плазми. Необхідним є регулярний контроль загального аналізу сечі (як при ремісії, так і — більш часто — під час загострення); проба С.С. Зимницького в умовах помірного обмеження вживання рідини в день збору сечі для оцінки концентраційної функції нирок. З метою виявлення структурних особливостей нирок (оцінка розмірів, контурів, товщини та однорідності паренхіми, форми та розмірів чашково–мискового апарату, склеротичних змін) найширше застосовується ультразвукове дослідження, а також комп’ютерна та магнітно–резонансна томографія нирок. Використовуються також, проте рідше, ніж раніше, радіонуклідні методи (ренографія та сцинтиграфія нирок, які дозволяють оцінити окремо функцію кожної з нирок, надають додаткову інформацію про особливості функціонування судинної системи нирок, обструкції сечовивідних шляхів, рефлюкси) та екскреторна уронефрографія (пов’язана з такими недоліками, як значна експозиція іонізуючого випромінення та йодовмісних контрастних речовин).
Лікування. При загостренні хронічного пієлонефриту необхідно проводити адекватну антибактеріальну терапію. У хворих старших за 18 років з неускладненим пієлонефритом віддалений прогноз, як правило, сприятливий, у переважній більшості випадків зберігається нормальна функція нирок і не розвивається АГ.
Лікування АГ в осіб із хронічним пієлонефритом здійснюється за загальноприйнятими принципами.
1.4. Полікістоз нирок (ПК)
Визначення та клінічна картина. В англомовній літературі для відмежування цього стану від інших форм кістозного ураження нирок застосовують термін «автосомно–домінантна полікістозна хвороба нирок». ПК — вроджене системне порушення з переважним ураженням нирок, нерідко із залученням печінки, підшлункової залози, рідше — головного мозку та інших органів. Може розвиватися в осіб обох статей. Розвиток ПК пов’язують із різними мутаціями (їх описано декілька сотень) одного з двох генів, що кодують утворення протеїнів поліцистину–1 та –2 (PKD1 та PKD2 відповідно), що регулюють функцію кальцієвих каналів у мембрані клітин тубулярного апарату та судин нирок, а також деяких інших тканин). Патологічний ген від батьків із ПК успадковується дитиною у 50 % випадків. Приблизно у 5 % випадків ПК мутації вказаних генів є спонтанними.
Для ПК характерна наявність, як правило, численних, білатеральних кіст у нирках, які після травматизації можуть містити кров або нагноюватися. У 80 % осіб із ПК виявляються кісти печінки, у 30–40 % — кісти підшлункової залози; у 8 % — аневризми інтракраніальних судин. Іноді виявляються кісти передміхурової залози, ураження серця у вигляді пролапсу мітрального клапана та/або дилатації кореня аорти, дивертикульоз товстого кишечника. Ураження нирок зазвичай є провідним у клінічній картині і визначає прогноз. Приблизно у 60 % осіб із ПК спостерігаються епізоди макрогематурії, тенденція до збільшення розмірів та кількості кіст, що призводить до прогресуючого симетричного збільшення розмірів нирок. Незважаючи на це, впродовж десятиліть функція нирок може зберігатись у межах нормальних значень, але з 30–40–річного віку починається її зниження, що часто супроводжується розвитком АГ. У західних країнах особи з ПК становлять 10 % хворих із термінальною стадією ниркової недостатності, що знаходяться на гемодіалізі.
Поширеність та характер АГ. Виявляють не менше ніж у 50–60 % хворих на ПК; АГ є одним із найчастіших клінічних проявів ПК. Розвиток АГ при ПК вважають важливим критерієм прогресування ураження нирок. Розвиток АГ можливий з дитячого віку.
Механізми розвитку АГ: мають значення такі фактори:
1) стиснення інтраренальних артерій кістами з розвитком ішемізації ниркової тканини, хронічною активацією ренін–ангіотензинової та симпатичної систем;
2) поступова втрата функціонуючої паренхіми нирок,
3) затримка натрію нирками.
Діагностика. Генетичне дослідження не використовується для скринінгового обстеження через низьку специфічність. Інструментальним методом вибору для скринінгу та динамічного контролю є ультразвукове обстеження (у дорослих дозволяє виявити кісти розміром > 1 см у діаметрі).
Критеріями діагностики ПК при ультразвуковому обстеженні нирок є:
1) дві або більше уні– чи білатеральних кіст у осіб віком < 30 років;
2) дві або більше кіст у кожній з нирок в осіб віком 30–59 років;
3) чотири або більше кіст у кожній з нирок у осіб віком > 60 років.
Додатковим критерієм діагностики є виявлення кіст у печінці та підшлунковій залозі. За необхідності в осіб із невеликими та нечисленними кістами може застосовуватись комп’ютерна томографія з внутрішньовенним підсиленням, яка дозволяє виявляти кісти > 0,3 см у діаметрі, але пов’язана з ризиком контраст–індукованої нефропатії.
В осіб без сімейного анамнезу ПК необхідним є проведення диференційної діагностики з іншими варіантами кістозного ураження нирок: 1) прості кісти (поодинокі, без прогресування); 2) набуті кісти (нечисленні, можуть розвиватися в осіб, які мають знижену функцію нирок); 3) автосомно–рецесивна полікістозна хвороба нирок (кісти численні, виявляються у новонароджених, дітей раннього віку).
Лікування. До цього часу відсутні лікарські засоби, що уповільнюють формування кіст і прогресування ПК. Лікувальні підходи залежать від особливостей клінічної картини. За умов нормальної функції нирок при відсутності АГ та інших клінічних проявів рекомендують спостереження з регулярним контролем АТ, рівня креатиніну, розмірів нирок (за даними ультразвукового обстеження).
Лікування АГ проводиться за загальноприйнятими принципами, описаними в розділі «Гломерулонефрит». При наявності макрогематурії — постільний режим, анальгетики, достатня гідратація для підтримки об’єму сечі в межах 2–3 л/добу (при відсутності протипоказань); за наявності епізодів макрогематурії в анамнезі рекомендовано уникати прийому антитромботичних препаратів і травматизації в ділянці живота та попереку. В осіб з епізодами сечової інфекції проводиться стандартна антибактеріальна терапія (перша лінія — фторхінолони). Особам із термінальною стадією ниркової недостатності показані діаліз і трансплантація нирки.
1.5. Гідронефроз, обструктивна нефропатія
Визначення та клінічна картина. Гідронефроз — патологічний стан, що характеризується дилатацією чашково–мискового апарату нирки, пов’язаний з порушенням відтоку сечі з нирки — повною або частковою обструкцією сечовивідних шляхів. Як синонім гідронефрозу застосовують термін «обструктивна нефропатія». Можливі причини гідронефрозу: 1) вроджені структурні порушення нирок, сечоводу, сечового міхура або уретри, які ускладнюють відтік сечі; 2) набуті зміни цих структур внаслідок травм, хірургічних втручань, опромінення та ін.; 3) компресії структур сечовивідних шляхів ззовні (судинами, пухлинами, у т.ч. при аденомі передміхурової залози, білатеральна компресія сечоводів при вагітності, ретроперитонеальному фіброзі та ін.); 4) обтурація просвіту сечовивідних шляхів конкрементами, згустками крові (особливо часто — у місці впадіння сечоводу в сечовий міхур); 5) різні види рефлюксу (особливо міхурово–сечовідний); 6) нейрогенний сечовий міхур. Клінічні прояви залежать від особливостей обструкції (гостра чи хронічна, часткова чи повна, одно– чи двостороння). Наприклад, гостра обструкція сечоводу конкрементом викликає появу інтенсивного різкого болю в попереку, також часто спостерігають дизурію, нудоту/блювання; в той же час поступовий розвиток гідронефрозу може не супроводжуватись клінічними проявами. Порушення відтоку сечі може збільшувати ризик утворення конкрементів, підвищує ймовірність розвитку/загострення сечової інфекції (клінічні прояви можуть включати дизурію, лихоманку, інтоксикаційний синдром, піурію, епізоди макрогематурії). Внаслідок дисфункції тубулярного апарату можливий також розвиток гіпонатріємії, гіперхлоремічного метаболічного ацидозу. Сумарна функція нирок може залишатися збереженою навіть при тяжкому однобічному гідронефрозі (контралатеральна нирка забезпечує компенсацію функції ураженої нирки). Зменшення об’єму сумарної функціонуючої ниркової паренхіми супроводжується зниженням функції нирок і розвитком АГ.
Поширеність і характер АГ. АГ не є частим клінічним проявом при гідронефрозі, особливо в осіб з однобічним характером ураження. Розвиток АГ частіше спостерігається при двобічному тяжкому гідронефрозі, як правило, співпадає у часі з етапом зниження функції нирок.
Механізми розвитку АГ: зменшення функціонуючої ниркової паренхіми, хронічна ішемізація тканини нирок з активацією ренін–ангіотензинової системи.
Діагностика базується на даних інструментальних обстежень: ультразвукове обстеження, спіральна комп’ютерна томографія, магнітно–резонансна томографія; достатньо широко застосовуються також екскреторна уронефрографія (для встановлення локалізації і особливостей обструкції), ретроградна пієлографія, цистоуретерографія (особливо для діагностики міхурово–сечовідного рефлюкса). В осіб із приступами сечокам’яної хвороби перевагу надають спіральній комп’ютерній томографії (вона дозволяє виявити до 99 % конкрементів у сечовивідних шляхах, тоді як при ультразвуковому обстеженні та екскреторній уронефрографії деякі з конкрементів не візуалізуються). При дослідженні сечі у частині випадків зміни можуть не виявлятися, в інших — виявляють гематурію (незмінені негломерулярні еритроцити, переважно в осіб із сечокам’яною хворобою); лейкоцитурію (за наявності сечової інфекції); зниження концентраційної функції нирок за даними проби С.С. Зимницького, збільшення рН сечі (при ураженні дистальних канальців).
Лікування. Важливим компонентом терапії є усунення обструкції та забезпечення дренування нирки. При цьому тактика визначається характером обструкції: 1) при гострій обструкції верхніх сечовивідних шляхів можливою є нефротомія; 2) при хронічній обструкції — стентування сечоводів; 3) при обструкції нижніх сечовивідних шляхів — катетеризація уретри або надлобкова пункція та катетеризація. В осіб із сечокам’яною хворобою може бути застосована літотрипсія; при аденомі передміхурової залози — відповідні оперативні та медикаментозні підходи. При загостренні сечової інфекції проводять антибактеріальну терапію.
Лікування АГ здійснюється за загальноприйнятими принципами.
1.6. Гіпоплазія нирки
Визначення та клінічна картина. Гіпоплазія нирки — вроджене порушення розвитку нирки зі зменшенням кількості нефронів та її розмірів. У більшості випадків спостерігається також порушення структури тканини нирки (дефекти канальцевого апарату, інтерстиціальний фіброз та ін.), що позначається як гіподисплазія нирки. У більшості випадків гіпоплазія нирки є компонентом спадкових синдромів, пов’язаних із мутаціями різних генів; зазвичай такі хворі мають численні мальформації різних органів та систем, АГ і зниження функції нирок (особливо при двосторонній гіпоплазії); діагноз часто встановлюють в дитячому, навіть у ранньому дитячому, віці. Гіпоплазія нирки може бути ізольованим вродженим порушенням; при односторонній локалізації за відсутності ішемізації ниркової тканини та збереженні сумарної функції нирок клінічні прояви можуть бути повністю відсутні; діагноз у таких осіб встановлюють випадково (наприклад, при скринінговому ультразвуковому обстеженні). АГ виникає, як правило, при наявності судинних порушень, що сприяють ішемії тканини гіпоплазованої нирки (наприклад, аномалій ниркових артерій, в тому числі — інтраренальних).
Поширеність та характер АГ. Її виявляють у 20–25 % дорослих осіб із гіпоплазією нирки; ступінь гіпертензії широко різниться.
Механізми розвитку АГ. В основі розвитку АГ у таких хворих лежить активація ренін–ангіотензинової системи внаслідок гіпоперфузії та ішемії нирки.
Діагностика. Припущення, що причиною АГ є гіпоплазія нирки, може виникнути, якщо виявляють зменшення розмірів її (нормальні розміри нирок у дорослих осіб: довжина — 100–155 мм, ширина — 50–70 мм, товщина 30–50 мм) за даними ультра–звукового, комп’ютерно–томографічного або інших інструментальних методів обстеження (екскреторна урографія, сцинтиграфія нирок). Таким пацієнтам необхідно провести стандартне загально клінічне та біохімічне лабораторні обстеження.
Лікування. За відсутності ефекту адекватної антигіпертензивної терапії, а також у випадках прогресуючого зниження функції нирок може знадобитися хірургічне втручання (реваскуляризація, нефректомія).
1.7. Ренінпродукуючі пухлини
Визначення та клінічна картина. У вузькому розумінні ренінпродукуючі пухлини — це пухлини юкстагломерулярного апарату нирок (синонім «реніноми»). Важливо зазначити (див. нижче розділ «Діагностика»), що підвищення активності реніну може спостерігатися і при інших пухлинах нирок (наприклад, нирково–клітинній карциномі, гемангіоперицитомі), а також метастатичних пухлинах інших локалізацій (легень, яєчників, печінки, підшлункової залози, саркомах, тератомах, парагангліомах та ін.). Підвищенню активності реніну в плазмі крові сприяють також великі інтраренальні пухлини, що здавлюють судинну систему нирки. Власне реніноми — це рідкі доброякісні пухлини, досить часто невеликого розміру (до 15–20 мм у діаметрі). Перебіг АГ на тлі реніном нерідко супроводжується вторинним гіперальдостеронізмом і пов’язаною з ним гіпокаліємією (інколи тяжкою, зі зниженням рівня калію крові в межах 2–3 ммоль/л), а також дуже високою активністю прореніну та реніну крові.
Поширеність та характер АГ. АГ виявляють у більшості пацієнтів із цією патологією, але описані поодинокі безсимптомні випадки; перебіг АГ, як правило, тяжкий і характеризується стабільно високим рівнем АТ, який погано піддається лікуванню; можливий кризовий перебіг захворювання. АГ зазвичай розвивається в осіб молодого віку (20–30 років), рідше — в середньому віці.
Механізм розвитку АГ: висока активність реніну крові.
Діагностика. Запідозрити наявність ренінпродукуючої пухлини можна, якщо у хворого молодого віку з тяжкою АГ і високим умістом (активністю) реніну в крові (нерідко — з вираженою гіпокаліємією) ураження судин нирок було виключене за даними артеріографії. Доцільним є проведення ультразвукового та комп’ютерного обстежень. При підозрі на наявність метастатичного ураження на тлі пухлин іншої локалізації проводяться відповідні обстеження. Остаточний діагноз може бути встановлено за даними морфологічного обстеження тканини видаленої пухлини, але в частині випадків за причини схожості гістологічної картини з такою первинних пухлин нирок заключний діагноз часто залишається не визначеним.
Лікування. Хірургічне видалення нирки з різним об’ємом втручання (часткова або радикальна нефректомія) призводить до усунення або значного зменшення тяжкості АГ та гіпокаліємії (як правило, протягом одного тижня після операції). У віддаленому післяопераційному періоді рецидивів чи метастазування реніном не спостерігається.
1.8. Травма нирки
Визначення та клінічна картина. Компресія ниркової паренхіми ззовні гематомою (внаслідок спортивної або іншої травми, а також ятрогенної природи (як ускладнення біопсії нирки), а також пухлиною чи кістою має визначення «синдром Пейджа» або «нирка Пейджа». Клінічна картина АГ внаслідок травми нирки може коливатися від безсимптомних або малосимптомних випадків до ситуацій з наявністю больового синдрому абдомінальної або поперекової локалізації, частіше — одностороннього характеру, макро– або мікрогематурії.
Поширеність та характер АГ. Підвищення АТ не є частим проявом травми нирки. Розвиток АГ можливий у випадках формування паранефральної або субкапсулярної гематоми і пов’язаної з нею компресії нирки. Частота АГ при тупій травмі нирки становить 2–10 %. АГ часто розвивається швидко, хоча можливий і віддалений її розвиток, у частині випадків погано контролюється медикаментозними засобами.
Механізм розвитку АГ: гіперактивація ренін–ангіо–тензинової системи внаслідок гіпоперфузії та ішемії тканини нирки.
Діагностика. В осіб з АГ незалежно від віку необхідно звернути увагу на наявність в анамнезі перенесених травм поперекової ділянки та/або живота (різної давності); встановити часовий зв’язок розвитку АГ із подібною травматизацією. Діагностичним методом вибору є комп’ютерна томографія нирок із внутрішньовенним контрастуванням.
Лікування. У частини пацієнтів прийнятним є вичікувальний консервативний підхід із застосуванням антигіпертензивних препаратів для контролю АТ, за показаннями — хірургічна евакуація гематоми.
1.9. Синдром Ліддла (Liddle)
Визначення та клінічна картина. Синдром Ліддла — рідкісне, генетично зумовлене (з автосомно–домінантним типом успадкування) порушення функції дистальних канальців нирки зі збільшенням реабсорбції натрію та вторинним посиленням секреції калію. В його основі лежать мутації генів SCNNIB або SCNNIG, що кодують бета– або гамма–субодиниці амілоридчутливих натрієвих каналів епітелію тубулярного апарату нирок.
Основні прояви синдрому Ліддла:
1) гіпокаліємія та гіперкалійурія;
2) артеріальна гіпертензія;
3) метаболічний алкалоз;
4) гіпернатріємія;
5) нормальний або знижений рівень альдостерону та реніну;
6) порушення інтелектуального та/або фізичного розвитку;
7) дегідратація, сонливість, м’язова слабкість, міалгії (як прояви гіпокаліємії).
Поширеність і характер АГ. АГ присутня практично в усіх хворих; зазвичай розвивається в осіб молодого віку (до 35 років), може бути діагностована в ранньому дитячому віці (перші роки життя); при відсутності лікування рівень АТ може бути досить високим.
Механізми розвитку АГ: хронічне перенавантаження натрієм та об’ємом. АГ при синдромі Ліддла є солечутливою та об’ємзалежною.
Діагностика. Запідозрити цей рідкісний синдром можна, якщо у хворого молодого віку (дитини раннього віку) АГ поєднується з переліченими вище порушеннями електролітного та кислотно–лужного балансу. Для підтвердження діагнозу рекомендується проведення генетичних обстежень.
Лікування. Обмеження кухонної солі в раціоні < 5 г/добу; калійзберігаючі діуретики (амілорид, тріамтерен), що блокують надмірно підвищену при синдромі Ліддла функцію натрієвих каналів тубулярного апарату нирок і призводять до зниження АТ і корекції гіпокаліємії та метаболічного алкалозу. Інші антигіпертензивні препарати (у т.ч. антагоністи альдостерону) малоефективні. При адекватному лікуванні прогноз є досить сприятливим. У частині випадків може бути застосована трансплантація нирки, після якої усуваються АГ та метаболічні порушення.
1.10. Синдром Гордона, або псевдогіпоальдостеронізм ІІ типу
Визначення та клінічна картина. Синдром Гордона — рідке генетично обумовлене (за автосомно–домінантним типом успадкування) порушення функції ниркових канальців. Залучена тіазидчутлива натрій–хлоридна котранспортна система у дистальних канальцях нефрона. Синдром обумовлений мутаціями двох різних, але взаємопов’язаних, генів, що кодують утворення білків, які беруть участь у регуляції натрій–хлоридної котранспортної системи.
Провідними клінічними ознаками синдрому Гордона є:
1) гіперкаліємія;
2) артеріальна гіпертензія;
3) метаболічний ацидоз;
4) нормальні показники швидкості клубочкової фільтрації.
Також можуть спостерігатися гіперхлоремія; гіпоренінемія при низьких або нормальних рівнях альдостерону; невисокий зріст; іноді — м’язова слабкість; в деяких випадках — порушення інтелектуального розвитку; порушення розвитку зубів.
Поширеність та характер АГ. Підвищення АТ має місце приблизно у 60–70 % осіб із цією патологією. АГ розвивається не відразу, у більшості випадків — на третьому десятилітті життя. За відсутності лікування рівень АТ може бути досить високим та відповідати 3–му ступеню АГ.
Механізми розвитку АГ: хронічне перенавантаження натрієм і об’ємом. АГ при синдромі Гордона — солечутлива і об’ємзалежна.
Діагностика. Припущення про наявність цього рідкісного синдрому може виникнути, якщо у молодого хворого АГ поєднується з переліченими вище порушеннями електролітного та кислотно–лужного балансу. Рекомендується визначення активності реніну та рівня альдостерону плазми. Доцільним є обстеження кровних родичів обох статей в різних поколіннях. Генетичні обстеження в широкій практиці досі недоступні.
Лікування. Обмеження кухонної солі в дієті; препарати вибору — тіазидні діуретики, їх застосування призводить до нормалізації АТ і рівня калію в крові, а відміна — до повернення цих, а також нових проявів захворювання. Позитивна відповідь на тіазидні діуретики зберігається роками/десятиліттями.
1.11. Реноваскулярна АГ
Варіанти уражень ниркових артерій, що призводять до розвитку реноваскулярної АГ, включають:
— фібром’язову дисплазію (ФМД);
— атеросклероз;
— системні васкуліти (артеріїт Такаясу, вузликовий поліартеріїт;
— розшарування аорти/ниркових артерій;
— тромбози при антифосфоліпідному синдромі, пухлинах та ін.;
— пошкодження судин нирок (травми при літотрипсії, трансплантації нирки, опромінювання й ін.);
— тиск ззовні (кістами, пухлинами, гематомами, при ретроперитонеальному фіброзі й ін.);
— артеріовенозні мальформації або фістули.
Поширеність і характер АГ. Реноваскулярні АГ становлять близько 1 % всіх випадків АГ. Серед осіб із рівнем діастолічного АТ ≥ 125 мм рт.ст. і наявністю ретинопатії ІІІ–ІV ступеня реноваскулярні АГ становлять до 30 % випадків. Оскільки найчастішою причиною реноваскулярних АГ є атеросклеротичне ураження ниркових артерій, поширеність їх зростає з віком. Характер АГ широко варіює: від безсимптомних форм і помірного підвищення АТ — до стійких високих цифр АТ (АГ 3–го ступеня), резистентної АГ з прогресуючим зниженням функції нирок.
Фібром’язова дисплазія — захворювання невідомої етіології (у 10 % хворих констатують генетичну схильність за автосомно–домінантним типом спадковості), характеризується фіброзуванням і/або надлишковою проліферацією клітин артеріальної стінки. Найбільш часто вражаються ниркові (до 75 % усіх випадків ФМД), сонні та хребтові артерії, рідше — мезентеріальні, плечові, клубові. Близько 1/3 осіб із ФМД мають ураження більше ніж одного судинного басейну. Зміни венозних та лімфатичних судин при ФМД відсутні. Поширеність АГ при ФМД становить 10–15 %. Середній вік встановлення діагнозу ФМД, за даними реєстру США, — близько 50 років, але часто (особливо при наявності раннього початку АГ) діагноз встановлюють у молодих дорослих чи у дітей. Частіше хворіють жінки. ФМД може призводити до потовщення стінок артерій зі зменшенням їх просвіту, також можливий розвиток ускладнень, включаючи формування аневризм і розшарування. Клінічна картина варіабельна: у частини пацієнтів ФМД перебігає безсимптомно, в інших випадках — симптоматика обумовлюється локалізацією і особливостями судинного ураження.
Варіанти клінічних проявів можуть включати:
1) при ФМД ниркових артерій — АГ, шуми над проекціями ниркових артерій;
2) при ФМД сонних і хребтових артерій — головні болі (за типом мігрені), запаморочення, болі в шиї, пульсуючі шуми в вухах, транзиторні ішемічні атаки/ішемічні інсульти, шуми над проекціями сонних артерій;
3) при ФМД мезентеріальних артерій — абдомінальний біль після їжі, зниження маси тіла;
4) при ФМД артерій кінцівок — біль у кінцівках при навантаженні, що зникає у спокої, різний АТ на кінцівках.
В осіб із ФМД ниркових артерій стенози частіше локалізовано в середніх і дистальних відділах судини, прогресування відмічається у 30–60 % пацієнтів упродовж 5 років.
Атеросклеротичне ураження становить до 90 % всіх випадків стенозів ниркових артерій. Серед осіб, яким проводиться катетеризація серця, поширеність атеросклеротичного ураження ниркових артерій досягає 30 %, а серед хворих, старших за 65–70 років, — 50 %. Типовою локалізацією судинного ураження при атеросклерозі є місце відходження від аорти ниркових артерій та їх проксимальна третина, у поєднанні з ураженням прилеглих відділів черевної аорти. При вираженому атеросклерозі ураження може мати дифузний характер із залученням інтраренальних артерій. Прогресування стенозу ниркових артерій впродовж 5 років спостерігається приблизно у 50 % пацієнтів. Поряд з АГ важливим клінічним проявом вважають зниження функції нирок (у зарубіжній літературі іноді використовують термін «ішемічна нефропатія»). Так, при обструкції > 60 % просвіту ниркової артерії спостерігається прогресуюче зменшення об’єму ниркової паренхіми і розвиток азотемії. Приблизно у 30 % хворих з атеросклеротичним ураженням ниркових артерій за відсутності лікування спостерігається зниження функції нирок протягом 6 років. За епідеміологічними даними, атеросклеротичне ураження ниркових артерій є причиною 14 % випадків термінальної ниркової недостатності, що потребує діалізного лікування.
Механізми розвитку АГ. Звуження ниркових артерій до 60–80 % просвіту при ФМД або склеротичному їх ураженні призводить до ішемії нирки, хронічної активації ренін–ангіотензинової і симпатичної систем, периферичної вазоконстрикції, затримки натрію і води. При тривалому істотному звуженні ниркових артерій розвиваються необоротні зміни мікроциркуляторного русла нирок, тубулоінтерстиціальний фіброз і гломерулосклероз.
Діагностика. Підходи до діагностики стенозів ниркових артерій викладені в Рекомендаціях American College of Cardiology Foundation/American Heart Association з нагляду за пацієнтами з ураженням периферичних артерій (2011). Рекомендовано такі методи обстеження:
— дуплексна ультрасонографія ниркових судин — скринінговий тест, що дозволяє оцінити розміри нирок, функціональний резерв кровотоку і розрахувати резистивний індекс; при оцінці гемодинамічно значимого (суттєвого) стенозу чутливість методу становить 92 %, специфічність — 85 %.
— комп’ютерно–томографічна ангіографія — скринінговий тест; зважаючи на ризик розвитку контраст–індукованої нефропатії (особливо при значному зниженні функції нирок), проведення його потребує обережності; результати дослідження складно інтерпретувати при наявності виражених кальцифікуючих уражень судин; чутливість для діагностики стенозу ниркових артерій — до 94 %, специфічність — 60–90 %;
— магнітно–резонансна ангіографія — скринінговий тест; застосовується з обережністю з огляду на ризик нефротоксичної дії гадолінійутримуючого контрасту у хворих із помірним/важким зниженням функції нирок; протипоказанням для застосування є імплантовані прилади різного роду (штучні водії ритму, кардіостимулятори та ін.); чутливість та специфічність у діагностиці стенозу ниркових артерій досягають 90–100 %;
— за недостатньої інформативності перелічених неінвазивних тестів та високої ймовірності стенозу — катетерна ангіографія ниркових артерій.
Не рекомендується використовувати скринінгові тести для діагностики стенозу ниркових артерій:
— каптоприловий тест із скануванням нирок;
— селективне визначення активності реніну в крові, що відтікає від ниркових вен;
— активність реніну плазми;
— каптоприловий тест з оцінкою активності реніну плазми після прийому каптоприлу.
Обстеження з метою виявлення клінічно значимого стенозу ниркових артерій показане в першу чергу таким категоріям хворих на АГ:
— із розвитком АГ у віці до 30 років;
— розвитком тяжкої АГ, що визначають як рівні систолічного АТ > 160 мм рт.ст. та/або діастолічного АТ > 110 мм рт.ст. віком 55 років і більше;
— раптовим та стійким погіршенням контролю АТ, який раніше добре піддавався лікуванню;
— резистентною АГ;
— злоякісною АГ
— значним погіршенням функції нирок на тлі прийому інгібіторів АПФ або сартанів;
— зменшенням розміру нирки (атрофія), а також різницею в розмірах нирок, що перевищує 1,5 см;
— епізодами раптового набряку легенів неясної етіології, особливо у хворого зі зниженою функцією нирок.
Пацієнтам зі встановленим діагнозом ФМД однієї локалізації рекомендуються неінвазивне обстеження інших судинних басейнів на предмет мультифокального артеріального ураження. Хворим із ФМД сонних або хребцевих артерій також рекомендується проведення ангіографічного обстеження інтракраніальних артерій для виключення наявності їх аневризматичних змін (при їх виявленні потрібні відповідні лікувальні підходи).
Лікування
Фібром’язова дисплазія. При безсимптомному перебігу та відсутності АГ стан вважається доброякісним, специфічне лікування не проводиться, рекомендується інструментальний контроль у динаміці в районі судинного ураження. Для зниження ризику тромбозів у таких пацієнтів може застосовуватись антитромбоцитарна терапія (ацетилсаліцилова кислота 75–100 мг/добу).
При наявності АГ для її контролю можна використовувати різні антигіпертензивні препарати (найбільш вивчені інгібітори АПФ або сартани, можливо в комбінації з блокаторами кальцієвих каналів та/або діуретиками), при цьому дуже важливий ретельний лікарський контроль і регулярна оцінка стану функції нирок.
Блокатори ренін–ангіотензинової системи протипоказані у випадках:
1) білатерального ураження ниркових артерій;
2) унілатерального ураження високого ступеня;
3) ураження судин єдиної нирки;
4) атрофії контралатеральної нирки;
5) ШКФ < 30 мл/хв/м2.
За відсутності ефекту від антигіпертензивної терапії, а також при тенденції до зниження функції нирок та розвитку інших ускладнень перевагу віддають черезшкірній балонній ангіопластиці, частіше без стентування, однак за наявності уражень або при надмірній податливості судинної стінки можуть використовуватись стенти.
Атеросклеротичне ураження ниркових артерій. Стандартне лікування включає:
1) багатокомпонентну антигіпертензивну терапію з використанням тих самих препаратів і з урахуванням тих самих пересторог, що й при лікуванні АГ на тлі ФМД;
2) застосування статинів у високих/максимальних дозах (цільовий рівень холестерину ліпопротеїнів низької щільності < 1,8 ммоль/л, в разі недосягнення цільового рівня — зниження на 50 % від вихідного вмісту в крові);
3) відмова від паління;
4) оптимальний контроль глікемії;
5) призначення ацетилсаліцилової кислоти в дозі 75–100 мг/добу після досягнення цільового АТ.
Показання до реваскуляризації (ангіопластика зі стентуванням) після публікації результатів масштабних досліджень ASTRAL і CORAL у таких пацієнтів значно звузились. У сучасних міжнародних рекомендаціях зазначається, що навіть у осіб зі зниженою функцією нирок (без швидкого прогресування за останні 6–12 міс.), протеїнурією > 1 г/добу, атрофією нирки і дифузним ураженням інтралатеральних артерій перевагу слід віддавати медикаментозній терапії за умов досягнення адекватного контролю АТ.
Натепер реваскуляризація є резервним методом лікування у таких випадках: 1) рецидивуюча та незрозуміла гостра серцева недостатність або набряк легенів; 2) рецидивуюча нестабільна стенокардія; 3) прогресуюче зниження функції нирок; 4) недостатня ефективність медикаметозної антигіпертензивної терапії або її непереносимість.
Хірургічне лікування (реваскуляризація) використовується значно рідше, переважно в осіб, яким неможливо виконати ангіопластику зі стентуванням, або при їх недостатній ефективності.
1.12. Довідкові матеріали
Викладення матеріалу в частині Рекомендацій, присвяченій диференційній діагностиці гіпертонічної хвороби і ниркових гіпертензій, базується на кількох опублікованих у 2012 і 2013 рр. міжнародних рекомендаціях KDIGO (Kidney Disease Improving Global Outcomes), до яких включені Рекомендації з діагностики і лікування хронічних захворювань нирок, Рекомендації з контролю АТ у хворих із хронічними захворюваннями нирок, Рекомендації з діагностики і лікування гломерулонефритів та ін.
Наводимо сучасну номенклатуру та принципи класифікації хронічних захворювань нирок і гострого ураження нирок.
Номенклатура і класифікація уражень нирок (KDIGO, 2012)
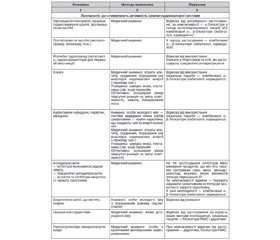
Хронічна хвороба нирок (ХХН). Цей термін натепер є загальноприйнятим. Під ХХН розуміють порушення структури та/або функції нирок, що наявне протягом > 3 місяців (табл. 1).
/135/135.jpg)
ХХН — це збірне поняття, що об’єднує гетерогенні захворювання, які порушують структуру та/або функцію нирок; мають різні клінічні особливості і неоднорідні за етіологією, ступенем тяжкості та темпом прогресування.
На різних етапах ХХН становлення і розвиток хвороби часто перебігають безсимптомно; їх діагностика при цьому зазвичай відбувається під час скринінгових або випадкових обстежень, а також при обстеженні з приводу супутніх патологічних станів. При швидкопрогресуючому перебігу деяких варіантів ХХН розвиток значного зниження функції нирок можливий вже за декілька тижнів/місяців, однак у багатьох випадках темп прогресування значно повільніший (роки — десятиліття), а в окремих ситуаціях при багаторічному спостереженні за хворими чіткого прогресування ХХН виявити не вдається.
Гостре пошкодження нирок (ГПН) і ХХН. Хворі на ХХН на всіх стадіях мають підвищений ризик розвитку ГПН (раніше використовували термін «гостра ниркова недостатність»).
Визначення ГПН (адаптовано за KDIGO–AKI, 2012)
ГПН визначається при наявності у хворого будь–якої з перелічених нижче ознак:
— підвищення рівня креатиніну сироватки крові ≥ 26,5 мкмоль/л протягом 48 годин;
— підвищення рівня креатиніну сироватки крові у 1,5 раза і вище порівняно з його вихідним значенням (або його ймовірним значенням — з урахуванням клінічної ситуації) впродовж останніх 7 днів;
— зменшення об’єму сечі до рівня < 0,5 мл/кг/год за 6 годин.
У більшості випадків під час тривалого перебігу ХХН на його тлі можуть розвиватися 1 епізод ГПН або більше. Диференційна діагностика між ГПН та ХХН при першому виявленні зниження функції нирок зазвичай базується на особливостях конкретної клінічної ситуації. Якщо у хворого зниження функції нирок (ШКФ < 60 мл/хв/1,73 м2) виявлено вперше під час якого–небудь гострого захворювання при відсутності даних про більш раннє ураження нирок, зазвичай припускають ГПН. Покращення функції нирок протягом декількох днів/тижнів може підтвердити діагноз ГПН у такого пацієнта. У той же час у хворого з аналогічним порушенням функції нирок, вперше виявленим, за відсутності гострого захворювання зазвичай припускають наявність ХХН. Збереження зниженої функції нирок при подальшому спостереженні є підставою для підтвердження ХХН. В обох випадках для встановлення більш точного діагнозу треба провести повторну оцінку стану функції нирок та маркерів ниркового ураження.
Класифікація ХХН. Експертами KDIGO (2012) запропоновано класифікувати ХХН за такими трьома характеристиками:
1) етіологія;
2) ШКФ;
3) рівень альбумінурії.
1. За етіологією виділяють 4 основні групи захворювань нирок:
1) гломерулярні;
2) тубулоінтерстиціальні;
3) судинні;
4) кістозні та вроджені ураження.
2. Класифікація за рівнем ШКФ наведена в табл. 2.
Якщо у хворого має місце ШКФ, що відповідає стадії І або ІІ, але немає маркерів ураження нирок (табл. 1), діагноз ХХН не встановлюють.
3. За рівнем альбумінурії та протеїнурії виділяють три основні категорії захворювань нирок:
— із нормальним або несуттєво підвищеним умістом альбуміну в сечі;
— із помірною альбумінурією;
— зі значною альбумінурією (табл. 3).
/136/136.jpg)
Протеїнурія/альбумінурія. Під терміном «протеїн–урія» розуміють наявність підвищеної кількості білка в сечі. Протеїнурія може бути наслідком втрати протеїнів плазми через:
— підвищення проникності клубочкового фільтру (альбумінурія, гломерулярна протеїнурія);
— недостатню канальцеву реабсорбцію профільтрованих низькомолекулярних протеїнів (тубулярна протеїнурія);
— підвищену концентрацію низькомолекулярних протеїнів у плазмі (протеїнурія переповнення, наприклад, з екскрецією легких ланцюгів імуноглобулінів при мієломі).
Протеїнурія також може відображати патологічну втрату білків, утворених безпосередньо в ниркових структурах (наприклад, у канальцях при їх пошкодженні) або в структурах сечовивідних шляхів (при інфекціях).
Ступінь вираженості протеїнурії розглядають як важливий фактор, що визначає темп прогресування ХХН. Є докази її безпосереднього пошкоджуючого впливу як на гломерулярні (подоцити — епітеліальні клітини капілярів клубочка), так і на тубулярні структури. Ступінь вираженості протеїнурії може бути використаний як один із маркерів прогнозування перебігу ХХН та відповіді на лікування.
Оцінка швидкості клубочкової фільтрації. ШКФ — найважливіший показник стану функції нирок. Основні методи визначення включають: 1) підрахунок з використанням формул на базі визначення рівня креатиніну сироватки крові (найбільш простий і дешевий, використовується повсюди); 2) підрахунок із використанням формул на базі визначення вмісту цистатину С сироватки крові; 3) безпосередня оцінка з використанням методик кліренсу (інуліну, ендогенного креатиніну та ін.).
З багаточисленних формул розрахунку ШКФ ми наводимо дві, що базуються на рівнях креатиніну сироватки крові — Кокрофта — Гаулта та MDRD. Ці формули вже достатньо добре відомі вітчизняним клініцистам і цілком придатні для використання в широкій практиці. Для ряду ситуацій більш точними можуть бути формули, запропоновані експертами KDIGO, 2012: CKD–EPI creatinine, що базується на визначенні креатиніну сироватки крові, та CKD–EPI сystatin C, що базуються на визначенні цистатину С сироватки (обидві ці формули дуже громіздкі, доступні онлайн www.kdigo.org).
А. Формула Кокрофта — Гаулта (при рівнях ШКФ < 10–15 мл/хв не використовується):
/135/135_2.jpg)
Б. Формула MDRD (особливо корисна при рівнях ШКФ < 10–15 мл/хв):
ШКФ = 170 x (Кр. x 0,0113) – 0,999 x вік (років) –– 0,176 x (Сечов. x 2,8) – 0,17 x Альб. x 0,762 (для жінок),
де Кр. — рівень креатиніну сироватки (мкмоль/л); Сечов. — рівень сечовини сироватки (ммоль/л); Альб. — рівень альбуміну сироватки (г/дл).
Розрахунок ШКФ за цією формулою можливий в online–режимі за допомогою пошуку в Google (ввести «калькулятор СКФ»).
2. АГ, зумовлена ендокринними захворюваннями
2.1. Первинний альдостеронізм
Первинний альдостеронізм (ПА, синдром Конна) — група станів, при яких продукція альдостерону клубочковою зоною кори надниркових залоз є непропорційно високою, повністю або частково автономною і не пригнічується при навантаженні натрієм.
Поширеність і характер АГ. Раніше ПА розглядали як рідкісну причину АГ (менше 1 %), у даний час він визнаний найбільш поширеною формою ендокринної симптоматичної артеріальної гіпертензії. Його частота в гіпертензивній популяції, за деякими даними, перевищує 10 %. Існує чітка залежність між тяжкістю АГ і частотою виявлення ПА: при рефрактерній АГ вона становить 17–23 %.
Механізми розвитку АГ. Останні десятиліття характеризуються переглядом ролі альдостерону у формуванні кардіоваскулярної патології. Добре відомі дані щодо його ролі в механізмах розвитку АГ — пригнічення синтезу реніну, затримка натрію, збільшення об’єму циркулюючої крові, акумуляція натрію в судинній стінці з підвищенням судинного опору і зростання її чутливості до вазоконстрикторних стимулів, розвиток низькоренінової гіпертензії — доповнилися даними щодо універсальної ушкоджуючої дії альдостерону на серцево–судинну систему. Доведені прозапальні, профібротичні, прооксидативні, прямі та опосередковані проаритмогенні ефекти альдостерону. Гіперальдостеронемія асоційована з самостійним, незалежним від АГ, підвищеним ризиком розвитку кардіоваскулярних ускладнень та смерті.
Клінічна картина. ПА об’єднує групу захворювань:
— альдостерон–продукуюча аденома — синдром Конна;
— ідіопатичний гіперальдостеронізм — двобічна гіперплазія клубочкової зони кори надниркових залоз;
— первинна однобічна гіперплазія кори надниркових залоз;
— сімейний гіперальдостеронізм I типу (пригнічується глюкокортикоїдами) і II типу (не пригнічується глюкокортикоїдами);
— альдостеронпродукуюча карцинома;
— альдостеронектопований синдром при екстраадреналовій локалізації альдостеронпродукуючої пухлини: щитоподібна залоза, яєчник, кишечник.
Превалюють альдостеронпродукуюча аденома та ідіопатичний гіперальдостеронізм — сумарно на їх частку припадає до 95 % випадків ПА. Частота альдостеронпродукуючої аденоми становить від 30 до 40 %, ідіопатичного гіперальдостеронізму — від 60 до 70 %.
Серед пацієнтів переважають особи середнього віку (30–50 років), більшість — жінки (60–70 %). Поодинокі випадки зустрічаються у дітей, переважно спадкового генезу.
Класична розгорнута клініка ПА — рідкісне явище, що зазвичай спостерігається при тяжких формах ПА. Вона описується серцево–судинним (АГ та її прояви), нейром’язовим (напади м’язової слабкості, судоми і паралічі переважно в ногах, шиї, пальцях рук) і нирковим (поліурією, ніктурією і полідипсією) синдромами.
Гіпокаліємія, частота якої становить від 9 до 37 %, тобто не більше ніж у третини хворих, на сьогодні не розглядається як обов’язковий діагностичний критерій ПА. Найбільш постійна ознака ПА — АГ. Діапазон тяжкості варіює від злоякісної, резистентної до м’якої АГ, високою є частота порушень циркадного ритму АТ (non–dipper, night–pickers). При сімейному гіперальдостеронізмі АГ має спадковий характер, маніфестує в ранньому віці.
Алгоритм діагностики
1. Виявлення пацієнтів із підвищеним ризиком ПА.
2. Визначення АРС — співвідношення концентрації альдостерону до активності реніну плазми крові.
3. При позитивному АРС — проведення одного з підтверджуючих тестів.
4. Встановлення типу ПА. При позитивному результаті підтверджуючого тесту проводиться КТ, субтест для встановлення типу ПА і виключення аденокарциноми. На цьому етапі приймається рішення (лікарем і пацієнтом) про медикаментозне чи хірургічне лікування.
5. Передопераційне обстеження. При двобічних ураженнях або непевних результатах візуалізаційних обстежень проводять вимірювання концентрації альдостерону та кортизолу в крові, що відтікає від надниркових залоз, — етап передопераційного обстеження для встановлення латералізації гормонпродукуючої пухлини. Це необхідно в зв’язку з відсутністю відповідності між розмірами пухлини та її активністю. При білатеральному ураженні, а також при відмові пацієнта від операції — лікування антагоністами мінералокортикоїдних рецепторів.
Виявлення пацієнтів із підвищеним ризиком ПА. Скринінгу підлягають такі категорії хворих: пацієнти з резистентною АГ, спонтанною або індукованою діуретиками гіпокаліємією, АГ і адреналовою інциденталомою (випадково визначеною пухлиною), АГ і сімейним анамнезом ранньої АГ або цереброваскулярних подій у віці до 40 років, гіпертензивні особи першого ступеня споріднення з хворими на ПА.
Визначення АРС (лабораторний скринінг). Вимірювання активності реніну плазми (або концентрації активного реніну — більш доступне в Україні) та рівня альдостерону, розрахунок АРС — визнаний тест первинного скринінгу ПА. Позитивним результатом вважають АРС понад 25 (20–30 або інше значення залежно від нормативів конкретної лабораторії) при вимірюванні реніну в нг/мл/год і альдостерону в нг/мл. При вимірюванні активного реніну плазми позитивним є співвідношення більше 6. Як додатковий критерій первинного діагнозу ПА враховується рівень альдостерону понад 15 нг/мл.
Підготовка до визначення АРС:
— корекція гіпокаліємії;
— споживання кухонної солі — без суворих обмежень;
— відміна прийому препаратів, що впливають на АРС принаймні за 4 тижні;
— спіронолактон, еплеренон, діуретики (в т.ч. калійзберігаючі), продукти лакриці, антигіпертензивні препарати (бета–блокатори, центральні a2–агоністи, інгібітори АПФ, АРА, інгібітори реніну, дигідропіридинові антагоністи кальцію), нестероїдні протизапальні препарати, оральні контрацептиви, естрогенні препарати.
Як антигіпертензивні засоби рекомендуються: пролонгований верапаміл, альфа–адреноблокатори, вазодилататори (гідралазин) у комбінації з верапамілом.
Процедура забору крові: протягом 2 годин пацієнт повинен знаходитись у вертикальному положенні (сидіти, стояти чи ходити). Безпосередньо перед забором крові о 8–9–й ранку необхідно 5–15 хвилин знаходитись в стані спокою, у положенні сидячи.
Інтерпретація значень АРС. Псевдопозитивні результати можуть бути одержані при нирковій недостатності, псевдоальдостеронізмі, в осіб старшого віку (> 65 років), при надмірному споживанні солей натрію і калію, прийомі бета–блокаторів, центральних a2–агоністів, інгібіторів реніну, нестероїдних протизапальних препаратів.
Псевдонегативні результати пов’язані: з прийомом діуретиків, інгібіторів АПФ і АРА, кальцієвих антагоністів, гіпокаліємією, низькосольовою дієтою, вагітністю, реноваскулярною АГ, злоякісною АГ, реніномою.
Підтверджуючі тести покликані встановити автономний характер секреції альдостерону. Як і подальші дослідження, вони проводяться в спеціалізованих закладах.
1. Внутрішньовенне введення NaCl: після 1–годинного перебування у положенні лежачи вводиться 2 л 0,9% розчину NaCl протягом 4 годин. До і після завершення тесту визначають рівень активного реніну, альдостерону, кортизолу, калію. Необхідний постійний моніторинг АТ і ЧСС.
Якщо альдостерон < 5 нг/дл — ПА малоймовірний, якщо альдостерон > 10 нг/дл — ПА високоймовірний: альдостерон 5–10 нг/дл — невизначений результат.
2. Супресивний тест з флудрокортизоном: 0,1 мг флудрокортизону протягом 4 днів приймається перорально кожні 6 годин з одночасним вживанням пролонгованих форм KCl (цільовий рівень K у крові близько 4 ммоль/л) і NaCl (30 ммоль тричі на день з прийомом їжі). Кров для вимірювання активного реніну та рівня альдостерону забирається о 10–й ранку, кортизолу — двічі — о 7–й і о 10–й годині в положенні пацієнта сидячи.
Альдостерон > 6 нг/дл підтверджує ПА при рівні активного реніну менше 1 нг/мл/ч і концентрації кортизолу більш високій о 10–й, ніж о 7–й ранку (для виключення ефектів АКТГ). У нормі альдостерон знижується < 30 %. При ПА рівень альдостерону залишається підвищеним, зберігається супресія реніну. Відмінність ідіопатичного гіперальдостеронізму полягає в тому, що порівняно з альдостеронпродукуючою аденомою концентрація альдостерону дещо знижується.
Сольове навантаження протипоказане пацієнтам з нирковою або серцевою недостатністю, порушенням ритму серця, вираженою гіпокаліємією.
Встановлення типу ПА і виключення аденокарциноми. Рекомендується КТ–дослідження з внутрішньовенним контрастуванням, а у дітей, вагітних, жінок, які годують груддю, — МРТ.
Передопераційне обстеження. При вирішенні питання про оперативне втручання проводиться визначення концентрації альдостерону в крові, що відтікає від надниркових залоз. Так званий селективний забір крові з надниркових вен є складною і дорогою ангіографічною процедурою, що вимагає великого досвіду лікаря–рентгенолога. Для виключення помилкових результатів проводиться одночасне визначення альдостерону і кортизолу в крові. Порівнюється не концентрація альдостерону з кожного боку, а співвідношення альдостерон/кортизол. Крім того, точність положення катетера в наднирковій вені підтверджується більш високою концентрацією кортизолу в даних пробах порівняно з нижньою порожнистою веною. Ця проба не є обов’язковою в осіб, молодших за 40 років, з однобічною альдостеронпродукуючою аденомою. Топічна і функціонально–топічна діагностика важлива для прийняття рішення про метод лікування, оскільки однобічна адреналектомія може не призвести до поліпшення або може супроводжуватись рецидивом хвороби в разі неадекватної оцінки результатів КТ або МРТ.
Лікування. При однобічному процесі (альдостеронпродукуюча аденома або однобічна гіперплазія) показана лапароскопічна адреналектомія. При білатеральному ураженні, а також при відмові пацієнта від операції — лікування антагоністами мінералокортикоїдних рецепторів.
Нормалізація рівня калію в крові після адреналектомії досягається практично в усіх пацієнтів. АГ виліковується при альдостеронпродукуючій аденомі у 50 % (35–70 %). Факторами, що асоціюються з ліквідацією АГ, є високі значення АРС та добової екскреції альдостерону, позитивна передопераційна відповідь на спіронолактон. Найчастіші причини збереження АГ після операції — супутня АГ невідомої природи, старший вік, тривала АГ.
У передопераційному періоді необхідний контроль артеріального тиску і концентрації калію в крові. Часто необхідне призначення антагоністів альдостеронових рецепторів.
У післяопераційному періоді визначається рівень альдостерону і активного реніну в крові. Скасовується прийом препаратів калію, спіронолактону, зменшуються (під контролем АТ) дози антигіпертензивних засобів. Зниження АТ відбувається протягом 1–6 місяців (іноді — до 1 року) після операції.
Поповнення ОЦК проводиться фізіологічним розчином. Препарати калію потрібні тільки при його рівні в крові менше 3 ммоль/л. Необхідна високосольова дієта, що визначається гіпоальдостеронізмом із ризиком гіперкаліємії у зв’язку з тривалою супресією контралатеральної надниркової залози. У поодиноких випадках потрібне тимчасове призначення флудрокортизону.
Медикаментозне лікування проводиться у випадку відмови пацієнта від операції, або при неможливості проведення оперативного втручання, або при двобічному процесі. З антагоністів мінералокортикоїдних рецепторів препаратом вибору є спіронолактон (верошпірон). Початкова доза 25 мг (12,5 мг) приймається під час їжі, титрується кожні 2 тижні під контролем АТ і рівня калію в крові. В даний час максимальною вважається доза 200 мг. Якщо розвиваються ускладнення (близько 7 % при дозі менше 50 мг і 52 % — понад 150 мг), пацієнт переводиться на еплеренон. Початкова доза еплеренону — 25 мг.
Хоча калійзберігаючі діуретики (насамперед амілорид як найбільш вивчений) не настільки ефективні, як антагоністи альдостерону, проте вони можуть бути, за необхідності, корисною альтернативою. Антигіпертензивні препарати (блокатори системи ренін–ангіотензину, антагоністи кальцію) знижують АТ, але не мають відчутного впливу на гіперпродукцію альдостерону.
При сімейному гіперальдостеронізмі I типу рекомендується з метою пригнічення секреції АКТГ призначати глюкокортикоїди (у дорослих — дексаметазон у початкових дозах 0,125–0,25 мг/добу, преднізолон — 2,5–5 мг/добу) перед сном у найменшій ефективній дозі під контролем рівня калію крові та АТ. У частини хворих для контролю АГ додатково потрібне призначення антагоністів альдостеронових рецепторів.
2.2. Феохромоцитома
Визначення та клінічна картина. Феохромоцитома, надниркова парагангліома, позанадниркова симпатична та парасимпатична парагангліома — нейроендокринні пухлини, що походять з адреналових хромафінних клітин або подібних клітин у симпатичних і парасимпатичних парагангліях. Феохромоцитома і симпатичні парагангліоми продукують, накопичують, метаболізують і секретують катехоламіни та їх метаболіти. Парасимпатичні парагангліоми рідко секретують значну кількість катехоламінів.
Клінічні симптоми захворювання обумовлені перманентним або постійним надлишком адреналіну і норадреналіну, у більш рідких випадках — допаміну. Існує залежність між біохімічним фенотипом і клінічним перебігом: надмірна секреція норадреналіну асоціюється зі стабільною АГ, адреналіну — з пароксизмальною та ортостатичною АГ, допаміну — з нормотензією. Відсутня чітка кореляція між розмірами пухлини, рівнем катехоламінів у крові і клінічною картиною, однак надзвичайно рідко клініка виникає при розмірах пухлини, менших за 2–3 см.
При різноманітності клінічних масок, що створили феохромоцитомі репутацію «великого імітатора» і утруднюють первинну діагностику, найбільш постійною і типовою є тріада: головний біль, серцебиття і пітливість (специфічність тріади — 94 %, чутливість — 91 %).
Власне феохромоцитома (80–90 % випадків) виникає в мозковій речовині надниркових залоз. Екстраадреналові симпатичні парагангліоми зазвичай розташовуються навколо нижньої мезентеріальної артерії та біфуркації аорти, рідше походять з хромафінної тканини черевної порожнини, малого таза, грудної порожнини. Екстраадреналові парасимпатичні парагангліоми в більшості випадків виявляються в ділянці голови та шиї. Позанадниркові катехоламінпродукуючі пухлини становлять близько 10 % усіх випадків. Злоякісні феохромоцитоми (феохромобластоми) зустрічаються нечасто (5–7 %).
Феохромоцитоми і парагангліоми належать до рідкісних пухлин: поширеність становить 1 : 65 000 — 1 : 250 000, хоча у великих автопсичних дослідженнях їх частота сягає 0,05–0,1 %. При стабільній АГ реєструються в 0,5–0,1 % хворих, що, ймовірно, становить лише 50 % від дійсної кількості, враховуючи, що у половини хворих АГ має пароксизмальний характер або наявна нормотензія.
Феохромоцитоми і парагангліоми мають подібні гістопатологічні характеристики. Їх розвиток у 24–27 % випадків (у дітей в 40 %) визначається відомими генетичними мутаціями. Феохромоцитоми можуть виникати спорадично або як складова спадкового синдрому. Спадкові пухлини часто асоційовані з множинною ендокринною неоплазією (МЕН) типу МЕН–2А або МЕН–2В, нейрофіброматозом 1–го типу (НФ 1), синдромом Хіппеля — Ліндау (СХЛ). Сімейні парагангліоми і феохромоцитоми розвиваються у зв’язку з мутацією генів, що кодують субодиниці B, C і D сукцинатдегідрогенази (SDHB, SDHC, SDHD). Успадкування здійснюється за автосомно–домінантним типом.
Спорадичні феохромоцитоми зазвичай поодинокі, сімейні форми — двобічні, мультицентричні. Феохромоцитома зустрічається в будь–якому віці, зазвичай у 25–50 років, частіше у жінок, 10 % — у дитячому віці, переважно у хлопчиків. Спорадичні форми проявляються у віці 40–50 років, сімейні — раніше за 30 років. При МЕН феохромоцитома поєднується з медулярним раком щитоподібної залози, з гіперпаратиреозом. Феохромоцитоми можуть супроводжувати неповні форми нейрофіброматозу, синдром Кушинга, у 17 % — холелітіаз.
Поширеність і характер АГ. АГ розвивається у 80–90 % хворих із феохромоцитомою: близько 50 % припадає на стабільну АГ, 45 % — пароксизмальну АГ і 5–15 % — нормотензію.
Пароксизмальна гіпертензія (45 %) характерна для пухлин, що продукують адреналін і особливо МЕН–2–асоційованих феохромоцитом. Типовими є кризи з різким підвищенням артеріального тиску, нервово–психічними, ендокринно–обмінними, шлунково–кишковими і гематологічними симптомами. Тривалість нападу — від декількох хвилин до декількох годин. У своїй більшості атаки непрогнозовані: їх частота коливається від декількох на день до одного за кілька місяців. Можуть провокуватись емоційним або фізичним стресом, пальпацією черевної порожнини, зміною положення тіла, їжею, багатою на тирамін, препаратами (b–адреноблокатори, нікотин, трициклічні антидепресанти, морфін, метоклопрамід, дроперидол, похідні фенотіазину).
Стабільна гіпертензія (50 %) тісно корелює з постійно високими концентраціями норадреналіну в плазмі; добовий, денний і нічний АТ значно вищий, ніж при пухлинах, що секретують адреналін. При цій формі значно частіше, ніж при пароксизмальній і нормотензії, спостерігаються ортостатична гіпотензія, постуральна тахікардія, що викликають запаморочення і синкопе.
Нормотензія (5–15 %) частіше має місце при сімейних формах, малих розмірах пухлин, при пухлинах, що секретують допамін. У зв’язку з відсутністю скарг пухлина може бути випадковою знахідкою.
Гіпертензія у дітей: рідкісною є пароксизмальна форма, 60–90 % дітей мають стабільну АГ, 20 % — нормотензивні. Приблизно у 80 % реєструється ортостатична гіпотензія. Симптоми феохромоцитоми дуже варіабельні, найбільш постійно відзначаються головні болі та пітливість.
Гіпертензія у вагітних: часто помилково діагностується прееклампсія. Феохромоцитома проявляється незалежно від термінів вагітності. Викид катехоламінів частіше пов’язаний зі змінами внутрішньочеревного тиску під час пологів, додатковою компресією при виношуванні плода, постуральними змінами. Гіпертензія має пароксизмальний характер. Недіагностована феохромоцитома може призвести до фатальних наслідків для матері та плода. Ризик їх смерті становить 50–60 %. З огляду на несприятливий прогноз видалення пухлини оптимально слід провести в перший–другий триместр або ж під час кесаревого розтину у разі виношування плода.
Алгоритм діагностики
1. Категорії хворих для скринінгу феохромоцитоми:
— АГ з пароксизмальним перебігом;
— швидкопрогресуюча або резистентна АГ;
— АГ з неадекватною реакцією на антигіпертензивні препарати;
— гіпертензія, що асоціюється з головним болем, пітливістю, блідістю або постуральними реакціями (гіпотензія і тахікардія);
— вказівки на феохромоцитому в сімейному анамнезі;
— інциденталома незалежно від наявності клінічних симптомів і АГ.
2. Лабораторні тести. Діагноз ґрунтується на виявленні надлишку катехоламінів. Продукти їх деградації — метанефрин і норметанефрин — можуть досліджуватися в плазмі та/або в сечі. Чутливість визначення вільного метанефрину в плазмі близько 96 %, специфічність — 85–100 %. З метою мінімізації хибнопозитивних результатів забір крові здійснюється в умовах спокою (через 8–12 годин після прийому їжі, напоїв із кофеїном, фізичного навантаження, паління) в положенні лежачи, через 15–20 хв після введення катетера. Визначення рівня метанефринів у сечі має таку ж чутливість, але більш низьку специфічність. Вимірювання метанефринів має незаперечні переваги над визначенням нативних катехоламінів. Чотирикратне перевищення верхньої межі норми асоціюється з 100% вірогідністю феохромоцитоми. Рівень метанефринів у плазмі, що перевищує верхню межу референтних значень, але менший, ніж 4–кратне підвищення, вимагає проведення супресорного тесту з клонідином.
Клонідин пригнічує секрецію адреналіну і норадреналіну мозковим шаром надниркових залоз у нормі, але не в осіб із феохромоцитомою. Методика тесту: за 10 хв до введення клонідину береться з вени кров для визначення концентрації катехоламінів. Потім хворий приймає 0,3 мг клонідину перорально і через 3 год знову проводиться забір крові. В осіб без феохромоцитоми після введення клонідину вміст норадреналіну і адреналіну в сироватці крові знижується порівняно з вихідними даними. У пацієнтів із феохромоцитомою зниження не відбувається.
3. Топічна діагностика. Спочатку проводиться КТ (у дітей, вагітних, жінок, які годують груддю, — МРТ) черевної ділянки: їх чутливість і специфічність становить 90–100 і 70–80 %. Екстраабдомінальне розташування парагангліїв вимагає МРТ–сканування всього тіла. Другим кроком топічної діагностики є сцинтиграфія з I131–метайодбензилгуанідином: метод високочутливий і специфічний, виявляє в надниркових залозах утворення, недоступні КТ та МРТ; обмеження пов’язані з низькою інформативністю щодо позанадниркових пухлин і метастазів. Третій крок: позитронно–емісійна томографія (ПЕТ), в ідеалі в поєднанні з КТ (ПЕТ/КТ) з використанням набору мічених лігандів. Він корисний, коли сцинтиграфія, КТ та МРТ не виявляють первинної пухлини, а також для локалізації метастазів. Четвертий крок: визначення катехоламінів в крові, що відтікає від надниркових залоз, використовується, коли методи візуалізації дають невизначені результати.
4. Генетичний аналіз. Класичні синдроми, що асоціюються з феохромоцитомою, МЕН–2, СХЛ і НФ 1–го типу, можуть бути підтверджені виявленням відомих генетичних мутацій. Також ідентифіковані мутації всіх 4 субодиниць сукцинатдегідрогенази, які є однією з найбільш частих причин спадкових парагангліом і феохромоцитом.
Найбільш небезпечні щодо злоякісності мутації сукцинатдегідрогенази — 30–60 % при SDHB. Ці пухлини поряд із норадреналіном продукують допамін, що дозволяє розглядати факт наявності підвищеного рівня допаміну в плазмі або сечі як маркер злоякісності процесу. Також при SDHB– і SDHD–мутаціях характерна екстраадреналова локалізація, що робить раціональним КТ або МРТ усього тіла. SDHD часто асоційована з розташуванням парагангліом у ділянці шиї і голови, де найбільш доцільним є метод МРТ–ангіографії.
Лікування — хірургічне. Рекомендується лапароскопічна техніка, яка в порівнянні з відкритою (трансабдомінальною, транслюмбальною) характеризується більш низькими показниками смертності, термінами госпіталізації, фінансовими витратами. У випадку двобічного ураження при спадкових формах можливе виконання субтотальної адреналектомії, щоб уникнути постійної замісної терапії. Класична лапаротомія (люмботомія) проводиться у випадках локально інвазивної або злоякісної феохромоцитоми.
Лікування АГ у період до операції: препаратом вибору є альфа–адреноблокатори для пригнічення ефектів циркулюючих катехоламінів (доксазозин). Показані антагоністи кальцію тривалої дії. Тахікардія, що вимагає додавання селективних бета–адреноблокаторів, в операційний період контролюється есмололом. Важливим компонентом лікування є корекція ОЦК.
Гіпертензивні кризи лікуються альфа–блокатором фентоламіном. Іншими препаратами вибору в ургентних станах є нітропрусид натрію, нітрогліцерин, урапідил, сульфат магнезії.
Оптимальним терміном підготовки до хірургічного лікування є терапія альфа–адреноблокаторами протягом 3–4 тижнів і більше, але можливі й екстрені оперативні втручання при неможливості забезпечити контроль гемодинамічних порушень. Ризик при таких втручаннях дуже високий.
У післяопераційний період необхідний ретельний контроль АТ, ЧСС, ОЦК, глікемії. Необхідно враховувати можливість рецидиву хвороби (5–14 %, для екстраадреналових пухлин — до 30 %).
2.3. Синдром Кушинга
Визначення та клінічна картина. Синдром Кушинга включає велику групу симптомів, що виникають у результаті тривалого впливу на тканини надлишкових концентрацій глюкокортикостероїдів. Розрізняють ендогенний і екзогенний (ятрогенний) синдром Кушинга. Надлишкова продукція кортизолу, характерна ознака ендогенного синдрому Кушинга, може розвиватися при підвищеній секреції адренокортикотропного гормона (АКТГ) первинною пухлиною гіпофіза (або ектопічною нейроендокринною пухлиною, що виробляє АКТГ) або в результаті АКТГ–незалежної гіперпродукції кортизолу наднирковими залозами. Таким чином, серед ендогенних причин гіперкортицизму виділяють хворобу Іценка — Кушинга (гіпофізарний АКТГ–залежний варіант), ектопічний АКТГ–залежний синдром Кушинга і власне синдром Кушинга — надниркового походження (аденома, карцинома, гіперплазія надниркових залоз).
Найчастішою причиною виникнення синдрому Кушинга є ятрогенний вплив. Клінічно виражений ендогенний синдром Кушинга зустрічається рідко — 2–5 нових випадків на 1 мільйон на рік. У переважної більшості (близько 70 %) причинним фактором є аденома гіпофіза — кортикотропінома. У той же час прихований (або субклінічний) синдром Кушинга, при якому може бути зареєстрована гіперпродукція кортизолу та відзначені окремі симптоми захворювання (АГ, ожиріння, цукровий діабет, остеопороз та ін.) може виявлятись досить часто (1–2 %) у популяції і ще з більшою частотою в певних групах пацієнтів. Цілеспрямований скринінг виявляє його у 5–9 % хворих із незадовільно контрольованим цукровим діабетом, у 0,5–1 % випадків АГ, у 10,8 % — у пацієнтів літнього віку з явищами остеопорозу та переломами хребців.
До характерних ознак синдрому Кушинга належать: повнокрів’я (плетора), місяцеподібне обличчя, багряно–червоні стриї шириною понад 1 см, проксимальна міопатія або проксимальна м’язова слабкість, у дітей — надмірна маса тіла і затримка росту, формування «бичачого горба» у верхній частині спини.
Асоційовані з синдромом Кушинга стани: АГ, остеопороз, остеопенія, цукровий діабет 2–го типу, синдром полікістозу яєчників, нефрокальциноз, гіпокаліємія, інциденталома надниркових залоз, схильність до інфекцій.
Поширеність і характер АГ. При ендогенному синдромі Кушинга АГ розвивається у 80 % пацієнтів, у той час як при ятрогенному — тільки в 20 %.
З огляду на низьку частоту синдрому Кушинга його діагностику рекомендується проводити за наявності характерних симптомів або після виключення інших причин вторинної гіпертензії. Важливість своєчасного виявлення та лікування пацієнтів із синдромом Кушинга визначається його значенням у формуванні високого рівня захворюваності та смертності. У разі вираженого гіперкортицизму середня виживаність становить 4,6 року, причина смерті — серцево–судинні ускладнення або інфекція. При цьому сучасні методи лікування дозволяють за умови нормалізації рівня кортизолу досягти рівня смертності, порівнянного з популяційним.
Алгоритм діагностики:
виключити використання глюкокортикостероїдів у будь–якій формі (у т.ч. у відбілюючих кремах, фітозборах і т.п.).
2. Ідентифікація пацієнтів, що підлягають скринінгу:
— із множинними і прогресуючими ознаками синдрому Кушинга, особливо патогномонічними (плетора, місяцеподібне обличчя, стрії, проксимальна міопатія);
— із нехарактерними для віку захворюваннями (остеопороз, АГ, цукровий діабет 2–го типу);
— інциденталома надниркових залоз;
— діти із затримкою росту і надмірною масою тіла.
3. Первинний скринінг. Для виявлення гіперкортицизму рекомендується проведення одного з 4 тестів:
— тест супресії з 1 мг дексаметазону — короткий (нічний) дексаметазоновий тест: дексаметазон приймають у дозі 1 мг на ніч о 23:00 або 00:00 з наступним вимірюванням рівня кортизолу крові о 8:00 або 9:00. Діагностичний критерій — концентрація кортизолу в сироватці понад 50 нмоль/л (18 нг/мл або 1,8 мкг/дл);
— дворазове визначення рівня вільного кортизолу в добовій порції сечі;
— дворазове визначення рівня пізнього вечірнього кортизолу в слині (перед сном або між 23:00 і 00:00). Верхня межа норми — 145 нг/дл (4 нмоль/л);
— тест супресії з 2 мг дексаметазону протягом 48 годин — довгий дексаметазоновий тест (рекомендується як тест другої лінії: процедура складна для амбулаторних умов, проводиться ендокринологом).
Перевага віддається нічному дексаметазоновому тесту.
Для первинного скринінгу не рекомендуються: вибіркове визначення рівня кортизолу в сироватці або АКТГ у плазмі крові, вимірювання 17–кетостероїдів у сечі.
У певних груп пацієнтів регламентовано проведення таких тестів для виключення синдрому Кушинга:
— у вагітних жінок — визначення концентрації кортизолу в сечі;
— у пацієнтів із тяжкою нирковою недостатністю — тест супресії з 1 мг дексаметазону;
— при інциденталомі надниркових залоз — тест супресії з 1 мг дексаметазону;
— при підозрі на циклічний синдром Кушинга — визначення екскреції кортизолу сечі і рівень пізнього вечірнього кортизолу.
4. Топічна діагностика. Найбільш прийнятний метод — спіральна мультидетекторна комп’ютерна томографія надниркових залоз із в/в контрастуванням, гіпофіза (метод вибору — високоенергетична магнітно–резонансна томографія з в/в контрастуванням і магнітним полем не менше 1,5–3 Тесла), ектопічної АКТГ–продукуючої пухлини — сцинтиграфія рецепторів соматостатину з 111In–пентетреотидом або позитронно–емісійна томографія з 18F–флюородеоксиглюкозою.
5. Категорії пацієнтів, які потребують подальшого обстеження в спеціалізованих закладах ендокринологічного профілю:
— зі збільшенням розмірів надниркових залоз;
— з аномальними результатами тестів;
— з початково нормальними результатами тестів при підозрі на циклічний гіперкортицизм або при появі з часом додаткових ознак гіперкортицизму;
— зі спадковими захворюваннями, пов’язаними з високим ризиком розвитку синдрому Кушинга (комплекс Карні, множинна ендокринна неоплазія–1 (синдром МЕН 1–го типу).
6. Другий, ендокринологічний, етап — підтвердження діагнозу синдрому Кушинга. У разі вираженого гіперкортицизму діагноз легко підтверджується повторенням тестів першої лінії. Ендокринологам рекомендується проводити такі тести:
— один або два із зазначених вище тестів першого ряду;
— рівень кортизолу сироватки опівночі (виявлення порушеного циркадного ритму);
— комбінований дексаметазон–кортикотропінрилізинг–гормон (декс–КРГ) тест.
Останній тест використовують для диференційної діагностики синдрому Кушинга і функціонального гіперкортицизму. Функціональний гіперкортицизм асоціюється з вагітністю, алкогольною залежністю, резистентністю до глюкокортикоїдів, вираженим ожирінням, незадовільним контролем цукрового діабету.
Інтермітуючий синдром Кушинга слід запідозрити за наявності клінічних ознак на тлі нормальних лабораторних тестів або навіть при минущому дефіциті кортизолу. Найпростіший спосіб встановлення цього діагнозу — попросити пацієнта зібрати зразки добової сечі або слини перед сном при повторному виникненні симптомів.
Лікування. Транссфеноїдальна аденомектомія — основний метод лікування хвороби Кушинга, що дозволяє досягти ремісії у 90 % випадків.
Однобічна адреналектомія показана при пухлині надниркової залози (глюкостерома, карцинома), а також при більш рідкісному варіанті АКТГ–незалежного надниркового синдрому Кушинга — АКТГ–незалежній макронодулярній гіперплазії надниркових залоз, первинній пігментній мікронодулярній хворобі надниркових залоз.
При стійкому післяопераційному гіперкортицизмі показана адреналектомія з використанням блокаторів стероїдогенезу: препарати мітотан або вітчизняний аналог хлодитан.
При тотальній і двобічній адреналектомії необхідна довічна замісна терапія глюкокортикостероїдами.
Для лікування АГ застосовують спіронолактон, калійзберігаючі діуретики, інгібітори АПФ, блокатори рецепторів ангіотензину ІІ, селективні бета–адреноблокатори.
2.4. Акромегалія
Визначення та клінічна картина. Акромегалія — нейроендокринне захворювання, що обумовлене значним підвищенням продукції соматотропного гормона (СТГ) передньої частки гіпофіза у дорослих і проявляється збільшенням розмірів кісток рук, стоп, нижньої щелепи та внутрішніх органів, а також порушеннями обміну речовин. При гіперпродукції СТГ у пубертатному віці у хворих розвивається гігантизм.
Головна причина акромегалії — пухлини гіпофіза (соматотропіноми), що автономно продукують СТГ. У 40–50 % хворих виявляються змішані аденоми гіпофіза, які продукують, крім СТГ, інші гормони: пролактин, тиреокортикотропний гормон, адренокортикотропний гормон, лютеїнізуючий гормон, фолікулостимулюючий гормон. Приблизно у 2 % хворих виявляються ектопічні пухлини, які розташовуються ендокраніально (глотковий та сфеноїдальний синус) і екстракраніально (підшлункова залоза, легені, середостіння).
Поширеність акромегалії становить 50–70 випадків на 1 мільйон населення за рік. Акромегалія виявляється в більшості випадків пізно — час від появи перших ознак до встановлення діагнозу перебуває в межах від 5 до 15 років. При нелікованій акромегалії відмічається висока смертність.
Для акромегалії характерні типові клінічні прояви: збільшення розмірів носа, губ, язика, нижньої щелепи, надбрівних дуг, збільшення кистей рук і стоп, потовщення шкіри, збільшення внутрішніх органів (спланхномегалія, зокрема кардіомегалія) у поєднанні з порушенням вуглеводного (цукровий діабет), мінерального (гіпокальціємія, вторинний гіперпаратиреоз) та ліпідного (дисліпідемія) обмінів, порушенням статевого розвитку (еректильна дисфункція, порушення менструального циклу). При акромегалії спостерігається дуже висока частота розвитку генералізованого атеросклерозу з переважним ураженням коронарних судин. Суттєвий внесок у розвиток ураження серця при акромегалії роблять метаболічні зміни в міокарді, мікроангіопатія, що обумовлені порушенням вуглеводного обміну. Всі ці процеси ведуть до прогресування атеросклеротичних, фіброгенних і дистрофічних змін у серці, розвитку порушень ритму та прогресування серцевої недостатності.
Поширеність і механізми розвитку АГ. Частота АГ при акромегалії становить 25–60 %. Причини АГ: затримка натрію і води в організмі, зниження продукції передсердного натрійуретичного пептиду та підвищення периферичного судинного опору внаслідок збільшення судинного тонусу. Характерним для акромегалії є ранній розвиток гіпертрофії лівого шлуночка з підвищенням серцевого викиду. Ці зміни пов’язуються з гіперпродукцією СТГ та його посередника — інсуліноподібного фактора росту–1 (ІФР–1).
Діагностика. Рентгенологічне обстеження черепа (типова деформація турецького сідла), КТ та МРТ голови та гіпофіза (візуалізація аденоми).
Лабораторна діагностика: а) визначення рівня СТГ у крові (у здорових СТГ ніколи не перевищує 10 мкг/л), який не знижується (щонайменше на 2 мкг/л) через 2 години після прийому 100 г глюкози; б) виявлення підвищеного рівня ІФР–1 у крові.
Лікування включає хірургічні, радіологічні та медикаментозні методи. Хірургічне лікування проводиться нейрохірургом (транскраніальна і транссфеноїдальна аденомектомія). У випадках, коли хірургічне лікування не показане або протипоказане, використовують променеву терапію (дистанційна гамма–терапія, протонотерапія, гамма–ніж). Медикаментозне специфічне лікування включає застосування аналогів соматостатину, агоністів дофаміну, антагоністів рецепторів гормона росту.
Медикаментозне лікування АГ не відрізняється від загальноприйнятих рекомендацій.
2.5. Тиреотоксикоз
Визначення та клінічна картина. Тиреотоксикоз — це клінічний синдром, що розвивається при захворюваннях щитоподібної залози і обумовлений надлишком тиреоїдних гормонів. Понад 80 % випадків тиреотоксикозу зумовлені дифузним токсичним зобом (автоімунне захворювання з гіперплазією та гіперфункцією щитоподібної залози). Окрім того, причинами тиреотоксикозу можуть бути вузловий (багатовузловий) токсичний зоб, гострий або підгострий тиреоїдит, медикаментозний тиреотоксикоз (при передозуванні препаратів тиреоїдних гормонів) та гормонально активна аденома гіпофіза (тиреотропінома).
Поширеність тиреотоксикозу в Україні, за даними статистики, становить 110 і більше випадків на 100 тисяч населення, хоча справжня частота становить не менше 1 % у жінок та 0,2 % у чоловіків.
Характерні клінічні прояви: дифузний або вузловий зоб, немотивована значна втрата маси тіла, пітливість, серцебиття, синусова тахікардія або фібриляція передсердь, емоційна лабільність, тремтіння рук, м’язова слабкість, екзофтальм та інші очні симптоми тиреотоксикозу. У тяжких випадках можливе виникнення застійної серцевої недостатності. У хворих із лімітованим коронарним резервом можливий розвиток синдрому стенокардії. Комплекс симптомів ураження серця отримав назву «тиреотоксичне серце».
Поширеність і механізми розвитку АГ. Частота АГ при тиреотоксикозі становить близько 30 %. Причина підвищення АТ — негативний вплив надлишку тиреоїдних гормонів на серцево–судинну систему, який проявляється в активації симпатоадреналової системи, підвищенні щільності і чутливості адренорецепторів (у першу чергу бета–адренорецепторів) до дії катехоламінів, що призводить до збільшення серцевого викиду, ЧСС, показників скоротливості міокарда, підвищення систолічного АТ із розвитком систолічної АГ. Водночас діастолічний АТ не підвищується, а, навпаки, може знижуватися внаслідок зменшення периферичного судинного опору завдяки вазодилатуючій дії трийодтироніну.
Діагностика. Головне значення має зниження рівня ТТГ — тиреотропного гормону гіпофіза (крім ТТГ — продукуючої аденоми гіпофіза) при підвищенні рівнів вільного тироксину (Т4) і вільного трийодтироніну (Т3). Відмічається підвищення вмісту в крові антитіл до рецептора ТТГ (при дифузному токсичному зобі).
При УЗД щитоподібної залози виявляється її дифузне збільшення або вузлові утворення.
Лікування тиреотоксикозу. Мета — стійка нормалізація рівня тиреоїдних гормонів. Методи лікування: а) консервативне: тиреостатичні препарати (тіамазол, карбімазол) до досягнення еутиреозу з подальшим терміном лікування до 18 місяців з наступним контролем через 12–24 місяці; б) хірургічне лікування (тиреоїдектомія); в) лікування радіоактивним йодом (в Україні майже не застосовується).
Лікування АГ повинно базуватися на бета–адреноблокаторах. Застосовуються селективні та неселективні бета–адреноблокатори. Останні вважають більш ефективними через те, що вони можуть впливати на синтез тироксину та трийодтироніну, зменшувати утворення трийодтироніну з тироксину. Можна застосовувати недигідропіридинові антагоністи кальцію, діуретики. Додатково призначають інгібітори АПФ.
2.6. Гіпотиреоз
Визначення та клінічна картина. Гіпотиреоз, або мікседема, — клінічний синдром, при якому спостерігається зниження функції щитоподібної залози, що може бути наслідком зниження секреції або доступності тиреоїдних гормонів для тканин організму.
Розрізняють первинний та вторинний гіпотиреоз. При первинному причиною є зміни у щитоподібній (щитовидній) залозі. При цьому рівень тиреотропного гормону гіпофіза (ТТГ) підвищений, а синтез тиреоїдних гормонів значно знижений. При вторинному гіпотиреозі патологічний процес локалізований у гіпоталамусі або гіпофізі. Вміст ТТГ знижений.
Поширеність гіпотиреозу в Україні — від 0,2 % серед чоловіків до 2 % — серед жінок. Запідозрити гіпотиреоз можна при наявності такого симптомокомплексу: сонливість, загальна та м’язова слабкість, сухість шкіри та слизових оболонок, збільшення маси тіла, облисіння, зниження ментальної функції, характерні набряки (при натисканні ямка не залишається, шкіра не збирається в зморшки, часто набряки переходять на слизові оболонки, що може призвести до зміни голосу та дикції), порушення нервової діяльності, включаючи зміни особистості. У більшості хворих підвищується діастолічний АТ, рівень якого корелює з тяжкістю гіпотиреозу. Пульсовий АТ нормальний або знижений. Часто виявляється синусова брадикардія, погіршення провідності серця. Порушення ліпідного обміну є передумовою швидкого та раннього розвитку атеросклерозу.
Поширеність і механізми розвитку АГ. Частота АГ при гіпотиреозі — 30–50 %. Існують дані про роль низки механізмів у розвитку АГ. Описане підвищення виключно діастолічного АТ при низькій активності реніну плазми крові. Встановлена роль периферичної вазоконстрикції в підвищенні діастолічного АТ. У свою чергу, периферична вазоконстрикція може виникати внаслідок зникнення вазодилатуючої дії трийодтироніну та зниження щільності бета–адренорецепторів, що призводить до стимуляції альфа–адренорецепторів. Наявні також дані про значення в підвищенні АТ активації симпатоадреналової системи та підвищення рівнів адреналіну і норадреналіну в крові.
Діагностика. Гормональне дослідження: при первинному гіпотиреозі — підвищення рівня ТТГ та зниження тиреоїдних гормонів (рівня вільного Т4 в крові). Гормональне обстеження дозволяє виявити субклінічний гіпотиреоз — підвищення ТТГ при нормальному рівні Т4. Діагностика субклінічного гіпотиреозу є дуже важливою в зв’язку з тим, що даний патологічний стан супроводжується низкою ускладнень, зокрема розвитком та прогресуванням серцево–судинних захворювань.
Лікування. Пожиттєва замісна терапія препаратами тиреоїдних гормонів (левотироксин). Дозу підбирають індивідуально. Мета терапії — підтримання рівня ТТГ у межах цільових значень 0,4–4,0 мкОд/мл (мМО/л).
Лікування АГ. Встановлено, що при досягненні стану еутиреозу відбувається нормалізація АТ. Перевагу віддають периферичним вазодилататорам — інгібіторам АПФ, дигідропіридиновим антагоністам кальцію, альфа–адреноблокаторам. Для підсилення антигіпертензивного ефекту застосовують діуретики.
2.7. Гіперпаратиреоз
Визначення та клінічна картина. Первинний гіперпаратиреоз — захворювання, що характеризується первинним підвищенням секреції гормону прищитоподібних залоз — паратиреоїдного гормона (ПТГ). Причиною найчастіше є аденома або гіперплазія прищитоподібної залози. Рідше спостерігається резистентність до ПТГ (псевдогіперпаратиреоз) або вторинний гіперпаратиреоз (при вираженому зниженні ШКФ та термінальній нирковій недостатності, надмірному вживанні (передозуванні) вітаміну D, кальцію, зниженні рівня фосфатів).
Поширеність гіперпаратиреозу — близько 0,3 % серед дорослого населення. Пік захворюваності — у 40–50 років. Жінки хворіють удвічі частіше, ніж чоловіки. Запідозрити гіперпаратиреоз можна при поєднанні АГ із гіперкальціємією. Клінічні симптоми неспецифічні: кістково–м’язові порушення (біль, деформації кісток, патологічні переломи, атрофія м’язів), нефрологічні порушення (рецидивуючий нефролітіаз), порушення з боку органів травлення (рецидивуюча виразкова хвороба шлунка і дванадцятипалої кишки, хронічний панкреатит, анорексія), зниження маси тіла, депресія, сонливість. Важливе місце в клініці гіперпаратиреозу займають АГ та порушення ритму серця.
Поширеність і механізми розвитку АГ. АГ виявлють у 20–70 % хворих на первинний гіперпаратиреоз. Гіперкальціємія та гіперпродукція ПТГ вважаються безпосередніми причинами систоло–діастолічної АГ. Показано, що при гіперпаратиреозі підвищується активність реніну плазми, під впливом ПТГ підвищується утворення АКТГ, внаслідок чого відбувається активація синтезу кортизолу і альдостерону, знижується чутливість судин до вазодилатаційних ендогенних субстанцій.
Діагностика. Вчасно встановити діагноз можна, лише проводячи скринінг кальцію крові. При виникненні підозри (за клінічними ознаками або на підставі випадкового виявлення гіперкальціємії чи підвищеного рівня ПТГ) необхідно:
1. Виключити вторинний характер (наявність ниркової недостатності, надмірне вживання вітаміну D, кальцію, в тому числі у складі біологічних добавок, полівітамінів.
2. Визначити рівень кальцію крові. Виявляється виражена гіперкальціємія за даними загального та іонізованого кальцію в крові. Референтні значення (нормальні рівні) загального кальцію в крові — 2,1–2,5 ммоль/л; іонізованого кальцію — 1,03–1,3 ммоль/л.
3. Визначити рівень ПТГ у крові (нормальний рівень ПТГ у крові — 20–70 пг/мл). Характерні гіпофосфатемія, підвищення лужної фосфатази в крові, гіперфосфатурія.
4. Інструментальне обстеження: а) візуалізація аденоми або гіперплазії прищитоподібної залози методом УЗ–діагностики, КТ, МРТ і радіонуклідних методів; б) виявлення кісткових порушень рентгенологічним методом, методом денситометрії та кісткової біопсії.
Лікування. Хірургічне лікування: видалення аденоми прищитоподібної залози або субтотальна паратиреоїдектомія при гіперплазії прищитоподібної залози. Медикаментозне лікування (дає тільки незначний частковий ефект): бісфосфонати, кальцитонін, замісна гормональна терапія, кальциміметики, вітамін D3. Хірургічне лікування приводить до нормалізації АТ у 20–100 % хворих.
Антигіпертензивна терапія проводиться за загальноприйнятими правилами. Деякі переваги мають діуретики, особливо салуретики, що зменшують електролітні порушення та утворення каменів.
2.8. Синдром полікістозних яєчників
Визначення та клінічна картина. Синдром полікістозних яєчників (синдром Штейна — Левенталя) — поліендокринний синдром, який супроводжується порушенням функції яєчників (відсутність або нерегулярність овуляції, підвищена секреція андрогенів та естрогенів), підшлункової залози (гіперсекреція інсуліну), кори надниркових залоз (гіперсекреція надниркових андрогенів). Назву «полікістоз яєчників» захворювання отримало завдяки наявності великої кількості кіст у яєчниках. Це фолікули, наповнені рідиною, що містять недоспілі яйцеклітини.
При полікістозі яєчників в організмі виробляється надмірна кількість андрогену, що призводить до гормонального дисбалансу. У цьому випадку овуляція відбувається рідше, ніж належить (олігоовуляція), або не відбувається взагалі (ановуляція). Припинення овуляції призводить до порушення або припинення менструації.
Причини полікістозу яєчників невідомі, але дослідження показують, що полікістоз яєчників пов’язаний із надлишком інсуліну — гормону, що виробляється підшлунковою залозою. Фактично синдром полікістозних яєчників — це класична модель стану інсулінорезистентності з усіма характерними для метаболічного синдрому ознаками (АГ, ожиріння, порушення обміну глюкози, дисліпідемія). Чималу роль у розвитку цього синдрому відіграють спадковість і генетичні фактори.
Поширеність АГ. АГ діагностується більше ніж у 50 % хворих на синдром полікістозних яєчників.
Діагностика. Запідозрити синдром полікістозних яєчників можна у жінок із АГ, що мають симптоми надлишку секреції андрогенів (гірсутизм та маскулінізація, андрогенна алопеція), порушення овуляції (оліго– або ановуляція, що є причиною безплідності), полікістозні яєчники при ультразвуковому дослідженні. Часто також спостерігаються: центральне ожиріння, порушення обміну глюкози (цукровий діабет, порушення толерантності до глюкози) та ліпідів (гіперхолестеринемія, гіпертригліцеридемія, зниження холестерину ліпопротеїнів високої щільності).
При підозрі на СПКЯ пацієнтка має бути направлена на обстеження до гінеколога. Кінцевий діагноз встановлюється гінекологом, гінекологом–ендокринологом при наявності двох із трьох указаних ознак (Роттердамські критерії, 2003):
1) порушення овуляції (оліго– і ановуляція);
2) прояви гіперандрогенемії — клінічні (гірсутизм, акне, себорея, алопеція) і лабораторні (підвищення рівня загального тестостерону і його вільної фракції);
3) виявлення ознак полікістозних яєчників за допомогою УЗД.
Діагноз синдрому полікістозних яєчників підтверджується при наявності двох із трьох діагностичних критеріїв (УЗ–ознаки полікістозу яєчників можуть і не виявлятися). Слід диференціювати з уродженою гіперплазією надниркових залоз із порушенням ферментації холестерину в кортикостероїди (характерним є підвищення 17–оксипрогестерону).
Лікування проводиться гінекологом–ендокринологом і включає:
1. Заходи, спрямовані на корекцію способу життя та зниження маси тіла.
2. Фармакологічні методи індукції овуляції.
3. Антиандрогенна терапія з метою лікування гірсутизму і акне.
4. Препарати, що підвищують чутливість тканин до інсуліну (метформін). Призначаються хворим із діабетом або предіабетом.
5. Хірургічне лікування.
Лікування АГ проводиться за загальноприйнятими правилами для хворих із порушенням вуглеводного обміну (ІАПФ, БРАІІ, антагоністи кальцію пролонгованої дії). Може застосовуватись моксонідин як препарат, що знижує інсулінорезистентність. Максимально виключаються препарати, що можуть посилити інсулінорезистентність (бета–адреноблокатори без вазодилатуючого ефекту). При ознаках затримки рідини призначаються діуретики без значних метаболічних ефектів (індапамід, торасемід).
3. Гемодинамічні АГ
3.1. Коарктація аорти
Коарктація аорти (вроджене сегментарне звуження аорти) виникає внаслідок дефекту аорти в процесі внутрішньоутробного розвитку плода. У дорослих причинами можуть бути: атеросклеротичне ураження, травма аорти, синдром дуги аорти (Такаясу), синдром Шерешевського — Тернера. Може локалізуватися проксимально від артеріального протоку, безпосередньо в місці відходження артеріального протоку або дистальніше за нього. Типовою (до 95 % випадків) є локалізація коарктації нижче відходження від аорти лівої підключичної артерії.
Поширеність і механізми розвитку АГ. АГ спостерігається практично в усіх хворих із коарктацією аорти, але підвищений АТ реєструється лише на верхніх кінцівках. Проксимально від місця звуження має місце артеріальна гіпертензія, дистально — гіпотензія. На ногах САТ на 50–60 мм рт.ст. нижчий, ніж на руках (100–110 мм рт.ст. або менше). Розвиток АГ пояснюють механічною перешкодою кровотоку в аорті, внаслідок чого лівий шлуночок, що нагнітає кров в аорту, постійно працює з перевантаженням. Компенсаторним механізмом є розвиток колатеральних судин, завдяки чому в нижню частину тіла деякий час потрапляє значна кількість крові, тому у дітей не реєструється висока гіпертензія. У період статевого дозрівання на тлі значного росту організму колатералі не в змозі забезпечити адекватне кровопостачання, і артеріальний тиск вище за місце коарктації аорти стрімко збільшується. Згідно з іншою гіпотезою, причиною АГ є ішемія нирок внаслідок зниження кровотоку в ниркових артеріях, що призводить до активації юкстагломерулярного апарату та включення ренін–ангіотензинового механізму.
Середня тривалість життя хворих із некорегованою коарктацією аорти становить 30–35 років.
Діагностика. Основа діагностики коарктації аорти — вимірювання тиску на обох руках і ногах (в підколінній ямці). Крім того, при огляді хворого визначається диспропорція м’язової системи верхньої та нижньої половини тіла (більш розвинуті м’язи плечового пояса, менше — стегон та ніг). Симптоми розвитку колатерального кровообігу проявляються підсиленою пульсацією артерій верхніх кінцівок та грудної стінки.
При ехокардіографічному дослідженні аорти у двохвимірному режимі візуалізується місце звуження аорти, при допплерівському дослідженні — турбулентний систолічний потік нижче за місце звуження. Ангіографія дозволяє встановити остаточний діагноз: виявити звуження аорти, значне розширення її висхідного відділу й лівої підключичної артерії, візуалізувати колатералі, через які ретроградно заповнюються міжреберні артерії.
Лікування. Медикаментозне лікування артеріальної гіпертензії у хворих із коарктацією аорти неефективне. Ця патологія є абсолютним показанням для хірургічного втручання, тому хворого необхідно направити на консультацію до кардіохірурга.
Після хірургічної корекції коарктації артеріальний тиск знижується поступово, протягом кількох років. У віддалений період (через 10–20 років) у 30–70 % випадках виникає рецидив артеріальної гіпертензії, частіше у хворих із тяжкою і тривалою гіпертензією до операції, тому хірургічне втручання повинно бути проведене якомога раніше.
Препаратами вибору для лікування АГ у хворих, які перенесли операцію з приводу коарктації аорти, є інгібітори АПФ, антагоністи кальцію, антагоністи рецепторів АІІ та блокатори бета–адренорецепторів.
3.2. Недостатність аортального клапана
Аортальна недостатність (неповне змикання стулок клапанів аорти в діастолу і регургітація крові з аорти в лівий шлуночок під час діастоли) може спричиняти підвищення систолічного АТ внаслідок збільшення серцевого викиду. Діастолічний тиск знижений, що є характерною ознакою цієї вади серця. Чим більша регургітація, тим нижчий діастолічний АТ. Пульсовий тиск підвищується і може досягати 80–100 мм рт.ст. Частота АГ у таких хворих перевищує 65 %.
Діагностика. Клінічні ознаки обумовлені коливаннями тиску в аорті й артеріальній системі. Уже дані загального огляду в разі вираженої аортальної недостатності дають змогу виділити характерні симптоми: патологічна пульсація сонних артерій (так званий танок каротид), артерій рук і ніг, пульсове розширення й звуження зіниць (ознака Ландольфі), ритмічне погойдування голови в такт пульсації сонних артерій (симптом Мюссе). Пульсація спричинена зміною кровонаповнення артеріол потужною пульсовою хвилею. Найвагомішою фізикальною ознакою аортальної недостатності є діастолічний шум над аортою, що виникає безпосередньо після II тону. У разі значного дефекту клапана II тон може зникати, а натомість прослуховується протодіастолічний шум.
Найчастішою ехокардіографічною ознакою вади є дилатація лівого шлуночка. Оцінювання регургітації слід проводити із застосуванням кольорового допплерівського обстеження, з оцінкою ступеня регургітації, розмірів висхідної аорти і vena contracta, ширини центрального потоку, що дозволяє вирішити питання про стадію захворювання і необхідність оперативного лікування.
Лікування АГ проводиться препаратами, що блокують РААС, і антагоністами Са. Застосування бета–блокаторів обмежене, оскільки вони подовжують діастолу і збільшують об’єм регургітації. Хворим з аортальною недостатністю III–IV стадій показане протезування аортального клапана. У віддаленому післяопераційному періоді АТ нормалізується в 40–50 % випадків, у 35–44 % хворих спостерігається підвищення діастолічного АТ і розвиток діастолічної гіпертензії.
4. Системні васкуліти
Системні васкуліти — це група захворювань, що характеризуються генералізованим ураженням судин за типом вогнищевого або сегментарного запалення і некрозу імунного генезу. Згідно з анатомічною класифікацією, розрізняють системні васкуліти з ураженням судин великого калібру (синдром дуги аорти (Такаясу), середнього та дрібного калібру (вузликовий періартеріїт); дрібного калібру (васкуліт Шенлейна — Геноха, гранулематоз Вегенера та інші).
4.1. Синдром дуги аорти
Визначення та клінічна картина. Неспецифічний аортоартеріїт (хвороба дуги аорти, хвороба Такаясу) характеризується гранулематозним запаленням аорти та її основних гілок із частковою або повною облітерацією та порушенням кровопостачання в тканинах та органах. Захворювання частіше зустрічається у жінок молодого віку (до 40 років). Клінічні прояви зумовлені порушенням кровообігу в різних органах, найчастіше — в кінцівках. Хворих турбує генералізований біль за типом міалгії, слабкість та парестезії в кінцівках, що посилюються під час фізичного навантаження, ознаки ішемічного ураження великих судин. Напади запаморочення можуть завершуватися втратою свідомості. Спостерігається тривала стійка субфебрильність, втрата маси тіла. Ускладнення захворювання проявляються кардіальною патологією — стенокардією, інсультом, аневризмою аорти, серцевою недостатністю. Можуть залучатися судини легенів, черевної порожнини, сітківки, що призводить до зниження або втрати зору.
Механізми розвитку АГ. Характерний симптом аортоартеріїту — артеріальна гіпертензія з різницею артеріального тиску на правій та лівій плечових артеріях більше ніж 10 мм рт.ст. або відсутність пульсу на одній або обох руках. Підвищення артеріального тиску має місце більше ніж у половини хворих, виникає внаслідок ішемії окремих сегментів нирок у результаті облітерації ниркових артерій у місці їх відходження від аорти, таким чином, АГ має вазоренальне походження.
Діагностика базується на клінічних проявах хвороби: асиметрія або відсутність пульсу, різниця в рівні артеріального тиску на руках, запаморочення, тимчасові порушення зору у молодих жінок. Діагноз верифікують за допомогою ангіографії. Найчастіше виявляють ураження підключичних артерій, низхідної аорти, ниркових та сонних артерій.
Лікування АГ проводиться за стандартною схемою. Доцільно якомога раніше виконати аортографію та при наявності показань провести реконструктивну операцію на судинах. В післяопераційному періоді хворому призначають кортикостероїди, на тлі яких артеріальний тиск зазвичай нормалізується.
4.2. Вузликовий періартеріїт
Визначення та клінічна картина. Вузликовий періартеріїт — системне судинне захворювання з утворенням аневризм артерій та вторинним ураженням внутрішніх органів. Хворіють переважно чоловіки віком 30–50 років, але захворювання зустрічається і у дітей, і у людей похилого віку. В основі лежить некротизуючий васкуліт артерій м’язового типу. Характерними є: гострий початок захворювання (іноді — поступовий) з підвищенням температури, болем у м’язах, швидкою втратою ваги, а також ураження нирок (у 80 % хворих) у вигляді нефропатії з помірним сечовим синдромом чи дифузного гломерулонефриту. У 43 % хворих виявляють ураження шкіри — вузлики, ліведо, пурпуру. Може спостерігатися ураження центральної нервової системи, що проявляється симптомами менінгоенцефаліту, моно– чи полінейропатіями. Для легеневого вузликового поліартеріїту характерна інтерстиціальна пневмонія, прогресуючий фіброз, інфаркт легенів, плеврит, астматичний синдром. У більшості хворих виявляється абдомінальний синдром: виразки шлунка та дванадцятипалої кишки та їх ускладнення у вигляді перфорації, перитоніту, шлунково–кишкових кровотеч. Ураження серця супроводжується приступами стенокардії, інфаркту міокарда внаслідок некротизуючого васкуліту.
Механізми розвитку АГ. За характером патогенетичних механізмів артеріальна гіпертензія є вазоренальною, оскільки артеріїт призводить до множинного внутрішньониркового стенозу артеріол із розвитком ішемії паренхіми нирки. Як відповідь на зниження перфузійного тиску в нирках збільшується секреція реніну з подальшим підвищенням артеріального тиску. Причиною артеріальної гіпертензії можуть бути також аневризми, розташовані біля біфуркації ниркових артерій, ускладнені розривом, інфарктами нирок. Ураження очей відображає ступінь тяжкості АГ: виявляють аневризми артерій, тромбози центральної артерії сітківки.
Діагностика. Характерним для вузликового періартеріїту є системний характер уражень. Найчастіший прояв захворювання — ураження нирок зі стійкою артеріальною гіпертензією, частіше — у вигляді нирково–абдомінального, нирково–серцевого, нирково–поліневритичного та інших системних синдромів. З метою виявлення аневризм або оклюзії вісцеральних артерій середнього калібру застосовується ангіографічне дослідження черевної аорти і судин, що відходять від неї. При біопсії шкіри та м’язів виявляють інфільтрацію артерій гранулоцитами та мононуклеарами.
/151/151.jpg)
/152/152.jpg)
Лікування. Ведення артеріальної гіпертензії у хворих із вузликовим поліартеріїтом передбачає контроль артеріального тиску та збереження функції нирок. Рекомендуються препарати, що зменшують активність ренін–ангіотензинової системи — інгібітори АПФ та блокатори рецепторів ангіотензину ІІ. Можна також призначати блокатори бета–адренорецепторів, при необхідності в комбінації з діуретиками. При застосуванні інгібіторів АПФ або БРАІІ необхідно контролювати функцію нирок.
5. АГ та синдром обструктивного апное сну
Визначення та клінічна картина. Синдром обструктивного апное–гіпопное сну (СОАС) — це стан, при якому у пацієнта виникають численні зупинки дихання, що повторюються, внаслідок повного (апное) або часткового (гіпопное) звуження дихальних шляхів під час сну на рівні глотки і припинення легеневої вентиляції при дихальних зусиллях, що зберігаються. Характеризується наявністю хропіння, зниженням рівня кисню в крові, грубою фрагментацією сну з частими пробудженнями і надмірною денною сонливістю. Пробудження при цьому є захисним механізмом, при якому відбувається активація м’язів — дилататорів верхніх дихальних шляхів і запобігає асфіксії. СОАС уражає 2–4 % загальної популяції, переважно чоловіків.
Причини СОАС:
— вроджене звуження дихальних шляхів;
— анатомічні дефекти на рівні носа та глотки (викривлення перегородки носа, поліпи, макроглосія, збільшення мигдаликів, низько розташоване м’яке піднебіння, подовжений піднебінний язичок, доброякісні та злоякісні новоутворення);
— вади розвитку лицьового черепа;
— звуження дихальних шляхів на фоні ожиріння;
— гіпотиреоз;
— акромегалія.
Поширеність і характер АГ. Частота артеріальної гіпертензії коливається від 35 до 80 % і залежить від ступеня респіраторних порушень. Характерною рисою артеріальної гіпертензії при синдромі обструктивного нічного апное є резистентність. При проведенні добового моніторування артеріального тиску в осіб з обструктивним нічним апное відзначається відсутність його нічного зниження. Під час зупинки дихання артеріальний тиск різко підвищується, тому вночі або вранці може виникнути гіпертонічний криз.
Діагностика
1. Анкетування з використанням діагностичних критеріїв та опитувальних шкал. Для оцінки специфічних симптомів і скарг використовуються спеціальні шкали. Найпоширенішою є Епвортська шкала денної сонливості.
2. Полісомнографічне дослідження є золотим стандартом у діагностиці СОАС. Воно включає одночасний запис:
— електроенцефалограми;
— електроокулограми;
— електроміограми;
— дихального потоку;
— насичення крові киснем;
— дихального зусилля;
— ЕКГ;
— положення тіла.
Частота епізодів обструкції верхніх дихальних шляхів під час сну визначається як «індекс апное + індекс гіпопное» (епізоди повного і часткового перекривання верхніх дихальних шляхів). Пацієнти з підозрою на наявність СОАС направляються у спеціалізовані центри, де є лабораторії полісомнографії для проведення цього дослідження.
3. Пульсоксиметричне моніторування під час сну. Портативний монітор веде запис повітряного потоку, дихального зусилля та насичення крові киснем. Виконується диференційна діагностика між епізодами обструктивного та центрального апное. Використовується для скринінгу пацієнтів на наявність СОАС.
Діагностичні критерії СОАС:
А. Присутній хоча б один із симптомів, перерахованих нижче:
1. Спонтанні засипання, денна сонливість, безсоння, нічний сон, що не освіжає, відчуття стомленості протягом дня.
2. Пробудження з відчуттям затримки дихання, дефіциту повітря або задухи.
3. Гучне хропіння та/або переривчасте дихання під час сну.
При інструментальному дослідженні виявляються:
Б. П’ять і більше будь–яких респіраторних епізодів обструктивного характеру за годину сну.
В. П’ятнадцать і більше будь–яких респіраторних епізодів обструктивного характеру за годину сну.
Г. Має місце симптоматика, не пов’язана з іншими захворюваннями і не обумовлена вживанням лікарських препаратів або психотропних речовин.
Для встановлення діагнозу СОАС необхідна наявність критеріїв А, Б і Г або В та Г.
Лікування. Пацієнтам із синдромом нічного апное рекомендують уникати спати в положенні на спині, якщо полісомнографічні дослідження демонструють порушення дихання саме в цій позиції. Необхідно знизити масу тіла за допомогою дієтичних, фармакологічних та хірургічних методів при ожирінні.
Стандартна антигіпертензивна терапія без відновлення прохідності дихальних шляхів малоефективна. Одним із основних способів лікування є застосування індивідуальних мікропроцесорних апаратів, що створюють постійний позитивний тиск у дихальних шляхах та забезпечують надходження повітря в легені пацієнта — терапія CPAP (Constant Positive Airway Pressure).
6. АГ, зумовлена неврологічними захворюваннями
6.1. Пухлини центральної нервової системи
Пухлини центральної нервової системи (ЦНС) розподіляються на первинні та вторинні (метастатичні). Первинні пухлини: внутрішньочерепні — в 90 % випадків та спінальні — в 10 %. Щодо мозку виділяють внутрішньомозкові (які розвиваються з клітин мозку) та позамозкові (розвиваються з оболонок мозку, черепних нервів, кісток черепа тощо). Вторинні (метастатичні) пухлини в більшості випадків бувають внутрішньомозковими і їх класифікують за локалізацією уражених часток мозку. Захворюваність на первинні та вторинні пухлини ЦНС становить 12–16 випадків на 100 тис. населення на рік.
Поширеність і механізми розвитку АГ. АГ виявляється при пухлинах ЦНС у більше ніж 30–40 % випадків. Однак цілеспрямованого виявлення АГ при пухлинах ЦНС не проводиться і точної статистики АГ немає.
У патогенезі АГ при пухлинах ЦНС головне місце відводиться підвищенню внутрішньочерепного тиску, порушенню мозкової ліквородинаміки, здавленню та/або руйнуванню мозкових структур у зонах мозку, що відповідальні за регуляцію АТ (довгастий мозок, гіпоталамічна ділянка), підвищенню тиску цереброспінальної рідини.
Діагностика. Виявлення характерних неврологічних симптомів:
а) локальні (симптоми подразнення: епілептичні припадки, галюцинації, локальні болі);
б) симптоми «віддалені» (вторинні, дислокаційні: частіше стовбурові, що обумовлені зміщенням і здавленням стовбурових відділів, ригідність м’язів потилиці, нудота, блювання, брадикардія, АГ, втрата свідомості);
в) загальномозкові (частіше внаслідок внутрішньочерепної гіпертензії: головний біль, нудота, блювання, пригнічення свідомості, застійні диски зорових нервів).
Наявність і комбінація симптомів обумовлена локалізацією, розмірами і гістологічною характеристикою пухлин.
Стандарти діагностики — МРТ головного мозку з внутрішньовенним контрастуванням. Можливе використання КТ (тривимірної спіральної) та за необхідності — селективної церебральної ангіографії, краніографії, електрофізіологічних методів, визначення пухлинних маркерів.
Лікування: хірургічне, застосування методів радіохірургії і радіотерапії, хіміотерапія, симптоматичне лікування.
Лікування АГ проводиться з урахуванням характеру підвищення АТ. Рекомендується застосування препаратів першої лінії з раціональним використанням діуретиків.
6.2. Енцефаліт
Енцефаліт — запалення речовини мозку. Виділяють первинні енцефаліти — самостійні захворювання, викликані нейротропними вірусами, мікробами і рикетсіями, і вторинні, що виникли на тлі основного захворювання: постекзантемні, поствакцинальні, бактеріальні й паразитарні та демієлінізуючі. Енцефалітам, що викликані нейротропними вірусами, притаманні епідеміологічність, контагіозність, сезонність і клімато–географічні особливості поширеності. Залежно від локалізації виділяють стовбурові, мозочкові, мезенцефальні та діенцефальні енцефаліти. При енцефалітах часто уражується не тільки головний, а й спинний мозок із розвитком енцефаломієліту.
Поширеність і механізми розвитку АГ. Частота стійкої АГ при різних формах енцефалітів — 15–20 %. Підвищення АТ є наслідком прямого ураження структур мозку, що відповідають за регуляцію АТ, а також результатом зростання внутрішньочерепного тиску і часто — тиску спинномозкової рідини. При енцефалітах спостерігається як стійка АГ, так і пароксизмальне підвищення АТ у вигляді симпатоадреналового пароксизму. АГ у більшості випадків асоціюється з вираженими вегетативними порушеннями.
Діагностика
1. Встановлення зв’язку з епідеміологічними і клімато–географічними особливостями та з вірусними й інфекційними захворюваннями.
2. Виявлення характерних клінічних ознак енцефаліту, що включають загальноінфекційні й вогнищеві та невогнищеві неврологічні симптоми. Специфічна імунологічна діагностика спрямована на виявлення збудника.
3. Інструментальна діагностика: МРТ, ехоенцефалоскопія, електроенцефалографія, УЗ–допплерографія, офтальмоскопія.
Лікування. Патогенетична терапія (дегідратація, десенсибілізація, глюкокортикоїди, антигіпоксанти, ангіопротектори, корекція водно–електролітного балансу, протизапальні засоби), етіотропна терапія (противірусні препарати), симптоматична терапія (антиконвульсантна, антипіретична терапія, лікування деліріозного синдрому, нормалізація сну), реабілітаційна терапія (лікування паркінсонізму, гіперкінезів, епілепсії Кожевникова, парезів та нейроендокринних порушень).
Лікування АГ. Використання препаратів першої лінії з урахуванням характеру АГ. Доцільне застосування бета–адреноблокаторів (за показаннями).
6.3. Травми мозку
Травми мозку — одна з головних причин смерті й інвалідизації населення. В Україні частота черепно–мозкових травм (ЧМТ) становить від 2 до 6 випадків на 1000 населения. При цьому 30–50 % хворих із тяжкою травмою помирають. Виділяють такі клінічні форми ЧМТ: струс мозку, удар мозку, дифузне аксональне ушкодження і здавлювання мозку.
Поширеність і механізми розвитку АГ. Частота АГ при ЧМТ — від 25 до 50 % і вище. АГ розвивається в 2 рази частіше, ніж гіпотензія. Частіше спостерігається тяжка АГ із високим АТ.
Причини АГ: механічні, судинні, порушення ліквородинаміки, нейроендокринні і психоемоційні фактори. Потужним механізмом розвитку АГ є виражена симпатоадреналова активація центрального генезу. Така АГ є стійкою і резистентною до терапії. У разі іритації вазомоторного центру стовбура мозку розвивається лабільна АГ. При ЧМТ часто спостерігається активація мінералокортикоїдної активності кори надниркових залоз, що викликає затримку натрію та надмірне виділення калію. Вказані зміни можуть бути причиною «відстроченої» АГ, що розвивається через 1–2 тижні після ЧМТ.
Діагностика. Дані анамнезу та характерні клінічні ознаки, обумовлені характером, локалізацією та фазою травми.
Інструментальне обстеження: МРТ, КТ, УЗ–дослідження мозку й інших органів (за показаннями), дослідження ліквору.
Лікування. Надання первинної допомоги, моніторинг та корекція внутрішньочерепного тиску, профілактика септичних ускладнень, протисудомна профілактична терапія, хірургічне лікування (за показаннями).
Лікування АГ: діуретики, ІАПФ, БРАІІ та інші препарати першої лінії з урахуванням особливостей перебігу АГ.
6.4. Спадкові порушення автономної регуляції
Вегетативна (автономна) нервова система (ВНС) представлена в корі великих півкуль головного мозку, гіпоталамічній ділянці, стовбурі мозку, спинному мозку та має периферичні відділи. Ураження вказаних структур проявляється вегетативними порушеннями. Натепер єдиної класифікації не існує. Виділяють центральні та периферичні вегетативні порушення, порушення ортостатичної толерантності, катехоламінові порушення та інші.
Порушення ВНС можуть бути викликані різними факторами (стресами, травмами, інфекціями, фізичними перевантаженнями, ендокринним дисбалансом, вагітністю й іншими). Але в основі цих порушень лежать спадкові дефекти вегетативної регуляції. У той же час серед порушень ВНС виділяються окремі патологічні стани і захворювання, що мають достатньо чіткі риси спадковообумовлених хвороб. До таких спадкових порушень ВНС належать такі.
Сімейна вегетативна дисфункція (СВД, або синдром Райлі — Дея) — рідка спадкова сенсорна вегетативна нейропатія. Виникає внаслідок мутації гена IKBKAP, що міститься на 9–й хромосомі. Тип спадковості — автосомно–рецесивний. Хворіють переважно євреї–ашкеназі. Носії генів захворювання серед євреїв–ашкеназі — 1 на 30 осіб, серед інших національностей — 1 на 3000 осіб. Захворювання характеризується широким спектром сенсорних дисфункцій і порушень функціонування ВНС, що викликані неповним розвитком сенсорних і вегетативних нейронів.
Поширеність і механізми розвитку АГ. АГ — один з основних клінічних симптомів. Має місце лабільний АТ, часті вегетативні кризи з підвищенням АТ на тлі ортостатичної гіпотензії. Причини АГ — порушення функціонування нейронів симпатичної і парасимпатичної ВНС.
Діагностика. Характерна клінічна картина. Захворювання проявляється з раннього дитинства. Основні симптоми: нечутливість до болю, відсутність сльозовиділення, затримка фізичного розвитку і здатності розмовляти, лабільний АТ, часті вегетативні кризи з підвищенням АТ. Діагностично вагомі критерії: національність — євреї–ашкеназі, відсутність грибоподібних сосочків на язику, зниження сухожилкових рефлексів, відсутність сліз при плачі.
Генетичне тестування — виявлення мутацій гена (точність — 99 %). Повинна проводитись пренатальна діагностика шляхом амніоцентезу або біопсії хоріона.
Лікування. Специфічних засобів не існує і хворі помирають у віці до 30 років. Симптоматичне лікування: бронходилататори за допомогою небулайзерів та інгаляторів, лікування пневмоній, діазепам, боротьба з ортостатичною гіпотензією (гідратація, дієта з достатньою кількістю солі, їжа малими порціями, тренування ніг), ортопедичні засоби, застосування штучної сльози.
Лікування АГ за допомогою антигіпертензивних препаратів першої лінії з урахуванням характеру АГ.
Мультисистемна атрофія та синдром Шая — Дрейджера — центральне вегетативне дегенеративне порушення, що проявляється вегетативною дисфункцією, синдромом паркінсонізму й атаксією в різних комбінаціях. При домінуванні вегетативної дисфункції використовується термін «синдром Шая — Дрейджера». Захворювання зараховується до рідкісних.
Поширеність і механізми розвитку АГ. АГ — один з основних симптомів. Частота АГ — до 90 %. Причини: порушення барорефлекторної регуляції. АГ виявляється в положенні лежачи і поєднується з ортостатичною гіпотензією. АГ при цьому захворюванні тяжка, АТ не знижується вночі і стає причиною інвалідності хворих.
Діагностика. Виявлення синдрому паркінсонізму, мозочкових знаків та вегетативної недостатності. Синдром паркінсонізму має ряд відмінностей від хвороби Паркінсона: ранні окорухові порушення, слабка відповідь на леводопу, фокальна дистонія, ранні поступальні порушення, феномен Рейно, дисфагія, насильні сміх та плач, контрактури. Вегетативна дисфункція проявляється ортостатичною гіпотензією, порушенням сечовиділення, імпотенцією. Наявні гастроінтестинальні симптоми: слинотеча, дисфагія, нудота. Ризик смерті високий у результаті пневмоній, легеневої емболії, інфекцій сечового тракту, раптової смерті.
Лікування симптоматичне. Лікування АГ із застосуванням препаратів першої лінії без використання діуретиків.
Ортостатична гіпотензія — порушення ортостатичної толерантності. Проявляється зниженням систолічного АТ більше ніж на 20 мм рт.ст. і діастолічного АТ на 10 мм рт.ст. у положенні стоячи, що супроводжується церебральною гіпоперфузією. Належить до рідкісних захворювань, виникає внаслідок дегенерації центрального та/або периферичного відділів ВНС.
Поширеність і механізми розвитку АГ. АГ виявляється у більшості хворих у положенні лежачи. Має місце тяжка АГ із відсутністю зниження АТ уночі. АГ у більшості випадків супроводжується тахікардією. Причина АГ — дегенерація цетральних і периферичних ланцюгів ВНС.
Діагностика. Виявлення ортостатичної гіпотензії, що супроводжується непритомністю. Для непритомності при ортостатичній гіпотензії характерні гіпо– і ангідроз («суха» непритомність) та відсутність зниження частоти серцевих скорочень. Характерна наявність тяжкої АГ у лежачому положенні, при якій АТ не знижується в нічний час.
Лікування. Високе положення голови, прийом їжі дрібними порціями, носіння еластичних гольфів, ізотонічні фізичні вправи, відмова від алкоголю і перебування на спекоті. Рекомендується вживання 3–4 г солі та до 3 л води.
Застосування мінералокортикоїдів (флудрокортизон), b–адреноміметиків (мідодрин), можливо — аналогів вазопресину, еритропоетину–b, кофеїну, йохімбіну, індометацину, антагоністів допаміну.
Лікування АГ проводиться обережно, без застосування діуретиків.
Синдром вегетативної дистонії (СВД) — симптомокомплекс, що проявляється перманентними ознаками симпатикотонії та/або парасимпатикотонії або кризами (пароксизмами). Може викликатися екзогенними (стреси, фізичне перевантаження й інше) та ендогенними факторами (ендокринні порушення — цукровий діабет, тиреотоксикоз, захворювання внутрішніх органів — панкреатит, виразкова хвороба шлунка і дванадцятипалої кишки), алергічні та нервові захворювання.
СВД переважно спадково обмовлений (конституційний). Виділяють такі варіанти синдрому: кардіоваскулярний, гіпервентиляційний, нейрогастральний та термоваскулярний. Поширеність СВД — до 30–40 %.
Поширеність і механізми розвитку АГ. Частота АГ при кардіоваскулярному варіанті синдрому — до 90 %. Причина АГ — дисфункція ВНС.
Діагностика. Характерні функціональні кардіоваскулярні, гіпервентиляційні, нейрогастральні та термоваскулярні порушення. При гіперсимпатикотонії: тахікардія, підвищення АТ, збліднення шкіри, відчуття страху при емоційному і фізичному навантаженні, послаблення перистальтики кишечника, мідріаз. При ваготонії: брадикардія, почервоніння шкіри, пітливість, зниження АТ, шлунково–кишкові дискінезії. Характерні кризи — симпатоадреналові, вагоінсулярні та змішані.
Лікування СВД із підвищенням АТ: седативні засоби, транквілізатори, бета–адреноблокатори. Можна використовувати ІАПФ, БРАІІ, антагоністи кальцію та агоністи імідазолінових рецепторів.
7. АГ, зумовлена медикаментами, наркотичними речовинами та харчовими компонентами
7.1. Кортикостероїди
Гормональні продукти кори надниркових залоз відповідно до їх основних ефектів розподіляють на глюкокортикоїди та мінералокортикоїди. В медичній практиці застосовують обидві ці групи. До глюкокортикоїдів зараховують: природні гідрокортизон ацетат, кортизон ацетат, кортизон та синтетичні — преднізолон, метилпреднізолон, дексаметазон, тріамцинолон та інші. До мінералокортикоїдних препаратів, що застосовуються у медичній практиці, належать синтетичні препарати — дезоксикортикостерону ацетат, дезоксикортикостерону триметилацетат (підставою для їх використання є хвороба Аддисона, міастенія, астенія, адинамія).
Медикаментозний екзогенний гіперкортицизм розвивається при лікуванні глюкокортикоїдами неендокринних захворювань. З метою досягнення клінічного ефекту призначають дози препаратів, що набагато перевищують фізіологічну потребу в них організму. Побічним ефектом є розвиток кушингоїдного симптомокомплексу.
В умовах тривалого прийому кортикостероїдів розвивається клінічна картина псевдогіперальдостеронізму із затримкою натрію в нирках і підвищенням об’єму циркулюючої крові, гіперкаліємічного метаболічного алкалозу. Гемодинамічно це проявляється збільшенням серцевого викиду, периферичного судинного опору й підвищенням артеріального тиску, погіршенням контролю артеріальної гіпертензії у хворих з уже існуючим анамнезом. Кортикостероїди сприяють підвищенню артеріального тиску також у результаті підвищення чутливості до норадреналіну й ангіотензину ІІ і збільшення судинної реактивності шляхом безпосереднього ефекту препаратів на тканину.
Найбільший ефект характерний для кортикостероїдів із мінералокортикоїдною активністю, таких як гідрокортизон і преднізолон. Проте деякі кортикостероїди з помірною мінералокортикоїдною активністю (дексаметазон, тріамцинолон) викликають невелику затримку натрію. Метилпреднізолон (метипред) не спричиняє затримки натрію.
Алгоритм дії лікаря. Необхідно ретельно зібрати анамнез, встановити наявність коморбідної патології та прийом хворим кортикостероїдів. Якщо змінити режим лікування неможливо, необхідно постійно моніторувати рівень натрію.
Лікування. Препаратами вибору для лікування АГ є діуретики, оскільки збільшення об’єму рідини є головним механізмом збільшення АТ у хворих, що довгостроково одержують кортикостероїди.
Синдром уявного надлишку мінералокортикоїдів може розвиватися через споживання великої кількості лакриці, що входить до складу жувальних гумок, кондитерських виробів або внаслідок прийому карбеноксолону. Причина — пригнічення 11–дегідрогенази активним алкалоїдом лакриці — гліциризиновою кислотою, що викликає блокування метаболізму кортизолу в нирках, стимуляцію мінералокортикоїдних рецепторів і розвиток мінералокортикоїдного синдрому. Для розвитку синдрому потрібна відносно невелика кількість лакриці — 50–100 г на день протягом 4 тижнів. Це викликає затримку натрію, посилений калійурез, підвищення АТ у раніше здорових людей, а також зниження активності реніну й рівня альдостерону в плазмі крові.
Алгоритм дії лікаря. При опитуванні хворого виявляються анамнестичні відомості стосовно вживання продуктів, що містять лакрицю, або прийому карбеноксолону.
Лабораторні показники: гіпокаліємія, зниження рівня альдостерону, гіпокаліємічний гіпоальдостеронізм, співвідношення «кортизол/кортизон» у сечі підвищене.
Для усунення артеріальної гіпертензії в такому випадку необхідно припинити вживання лакриці, після чого артеріальний тиск досить швидко нормалізується. Електролітні й гормональні порушення також оборотні.
7.2. Нестероїдні протизапальні препарати (НПЗП)
Встановлено вплив НПЗП на серцево–судинну захворюваність та смертність. Це обумовлено тим, що судинний гемостаз визначається балансом між тромбоксаном А2, який синтезується в тромбоцитах за участю ЦОГ–1 і підсилює їх агрегацію, та простацикліном, який синтезується в ендотелії судин за участю ЦОГ–2. При селективній блокаді ЦОГ–2 кількість тромбоцитів підвищується, що є ризиком тромбоутворення. НПЗП (за винятком ацетилсаліцилової кислоти) підвищують ризик інфаркту міокарда та інсульту. Напроксен є нейтральним щодо ризику кардіоваскулярних подій.
Усі НПЗП викликають затримку рідини за рахунок блокування натрійуретичного ефекту, обумовленого ЦОГ–2, а також вазоконстрикцію через блокаду простагландинів із вазодилататорним ефектом. Різні класи НПЗП відрізняються за своїм впливом на рівень артеріального тиску. Індометацин, піроксикам і напроксен асоціюються з найбільш вираженим підвищенням АТ. Аспірин не впливає не рівень артеріального тиску. Нові пероральні СОХ–2 інгібітори (коксиби) підвищують артеріальний тиск дозозалежним шляхом за типом традиційних НПЗП.
Алгоритм дії лікаря. Враховуючи наявність взаємозв’язку між ступенем кардіоваскулярного ризику та тривалістю лікування, рекомендується застосовувати НПЗП у низькій ефективній дозі та короткими курсами.
Згідно з рекомендаціями Європейської мультидисциплінарної групи експертів із використання НПЗП (2011), пацієнтам із високим кардіоваскулярним та низьким шлунково–кишковим ризиками допустимо використання напроксену в комбінації з інгібіторами протонної помпи. При поєднанні високого кардіоваскулярного та високого шлунково–кишкового ризиків рекомендується уникати призначення будь–якого препарату з групи НПЗП, а в разі клінічної необхідності допускається застосування інгібіторів ЦОГ–2 або диклофенаку/напроксену в комбінації з інгібіторами протонної помпи. Пацієнти з підвищеним кардіоваскулярним ризиком за відсутності протипоказань повинні приймати низькі дози ацетилсаліцилової кислоти.
Лікування. У хворих на артеріальну гіпертензію, які постійно одержують НПЗП, необхідно ретельно моніторувати АТ і вносити корекцію в режим антигіпертензивної терапії. Препаратами вибору є антагоністи кальцію й препарати центральної дії, оскільки їх антигіпертензивна активність не пов’язана з синтезом простагландинів.
7.3. Препарати жіночих та чоловічих статевих гормонів
Епідеміологічні дослідження демонструють, що в 5 % жінок, які довгостроково приймають пероральні протизаплідні препарати, розвивається симптоматична артеріальна гіпертензія. Дія на артеріальний тиск обумовлена тим, що високі дози естрогенів та прогестерону викликають затримку натрію й рідини за рахунок взаємодії з мінералокортикоїдними рецепторами або впливом на 11–гідроксистероїд–дегідрогеназу. Крім цього, при тривалому прийомі гормональних контрацептивів відбувається активація ренін–ангіотензинової системи, тому що естроген стимулює в печінці синтез ангіотензиногену.
Алгоритм дії лікаря. При наявності артеріальної гіпертензії рекомендується відміна контрацептивів, слід пропонувати альтернативні методи контрацепції. Препаратами вибору для лікування АГ є діуретики, при цьому артеріальний тиск нормалізується повільно — протягом 3 міс. і більше.
У жінок у постменопаузі з артеріальною гіпертензією також не рекомендується застосовувати комбінацію естрогену та прогестерону.
Тестостерон пропіонат: побічні ефекти, насамперед унаслідок застосування великих доз, обумовлені затримкою води та натрію, отже, провокують розвиток набряків, що особливо небезпечно для хворих із серцево–судинною патологією. У деяких осіб може реєструватися симптоматична артеріальна гіпертензія з проявами гіпертонічного кризу. Відміна гормональної терапії може призводити до нормалізації АТ у період від 2 до 12 місяців.
7.4. Антидепресанти
Тривалий прийом антидепресантів із групи інгібіторів моноаміноксидази здатний викликати артеріальну гіпертензію, у деяких випадках — зі значним підвищенням артеріального тиску. В основі гіпертензивної дії лежить блокування моноаміноксидази — ферменту, що викликає уповільнення інактивації моноамінів, у тому числі норадреналіну, серотоніну в центральній нервовій системі. Обумовлене цими препаратами накопичення в деяких структурах мозку вищезазначених нейромедіаторів відіграє основну роль у механізмах антидепресивного й гіпертензивного ефектів.
Алгоритм дії лікаря та лікування. Необхідно ретельно вивчити анамнез хворого. При гіпертензивних кризах, обумовлених застосуванням інгібіторів МАО, необхідно терміново використовувати b–адреноблокатор (фентолалін), що у цій ситуації є антидотом.
7.5. Рекомбінантний еритропоетин
Еритропоетин є перспективним препаратом для лікування анемії у хворих із нирковою недостатністю. Гіпертензивні ефекти проявляються підвищенням артеріального тиску на 10 % у 20–30 % гемодіалізних хворих. Артеріальна гіпертензія з’являється через 2–16 тижнів від початку лікування, є дозозалежною, не корелює зі зміною показників червоної крові. Потенційні механізми підвищення артеріального тиску: підвищена в’язкість крові, нейрогуморальна активація (симпатоадреналова і ренін–ангіотензинова системи), дисбаланс локальних вазоактивних агентів (підвищений синтез ендотеліну–1), мітогенний ефект, збільшення концентрації кальцію в цитозолі, ремоделювання судин, тобто прямі судинні ефекти.
Алгоритм дії лікаря та лікування. Необхідний регулярний контроль артеріального тиску у хворих, які лікуються еритропоетином. Артеріальна гіпертензія досить добре коригується. Препаратами вибору є антагоністи кальцію й блокатори бета–адренорецепторів. Діуретики, інгібітори АПФ, сартани менш ефективні, оскільки у хворих, яких лікували еритропоетином, об’єм крові не змінений, активність реніну плазми та рівень ангіотензину ІІ знижені. Якщо антигіпертензивна терапія неефективна, варто знизити дозу еритропоетину.
7.6. Імуносупресанти
Імуносупресивна терапія застосовується для лікування хворих, яким проведена трансплантація органа. Деякі хворі повинні постійно, довготривало приймати циклоспорин А, внаслідок чого може розвинутися нефротоксичний ефект зі зниженням гломерулярної фільтрації та підвищенням рівня креатиніну в крові; порушенням функції печінки та центральної нервової системи (тремор, нейропатія). Можливий розвиток артеріальної гіпертензії. Зазвичай має місце помірна артеріальна гіпертензія, але можливі варіанти тяжкої форми з енцефалопатією. Після припинення прийому препарату підвищений артеріальний тиск знижується, але повністю не нормалізується. Механізм виникнення артеріальної гіпертензії на тлі лікування циклоспорином пояснюється стимуляцією симпатоадреналової системи, затримкою натрію, посиленням синтезу і вивільненням ендотеліну.
Алгоритм дії лікаря та лікування. При лікуванні хворого циклоспорином необхідно контролювати АТ кожні 2 тижні протягом 3 місяців терапії і надалі продовжувати спостереження за хворим. При можливості дозу циклоспорину варто знизити на 25 % або замінити іншим імуносупресивним препаратом.
Антигіпертензивні препарати для лікування циклоспориніндукованої артеріальної гіпертензії: діуретики є ефективними, але вони можуть викликати порушення електролітного балансу, посилити азотемію; найбільш ефективні: антагоністи кальцію (але вони можуть підвищувати рівень циклоспорину в крові шляхом впливу на метаболізм цитохрому Р450), інгібітори АПФ; лабеталол і клонідин ефективні у деяких пацієнтів цієї групи.
Перелік скорочень
АГ — артеріальна гіпертензія.
АКТГ — адренокортикотропний гормон.
АПФ — ангіотензинперетворюючий фермент.
АРС — альдостерон–ренінове співвідношення.
АТ — артеріальний тиск.
БРА — блокатори рецепторів ангіотензину.
ВНС — вегетативна нервова система.
ГН — гломерулонефрит.
ГПН — гостре пошкодження нирок.
ДАТ — діастолічний артеріальний тиск.
КТ — комп’ютерна томографія.
МАО — моноаміноксидаза.
МЕН — множинна ендокринна неоплазія.
МРТ — магнітно–резонансна томографія.
НПЗП — нестероїдні протизапальні препарати.
НФ — нейрофіброматоз.
ПА — первинний альдостеронізм.
ПЕТ — позитронно–емісійна томографія.
ПН — полікістоз нирок.
ПТГ — паратиреоїдний гормон.
РААС — ренін–ангіотензин–альдостеронова система.
САТ — систолічний артеріальний тиск.
СВД — синдром вегетативної дистонії.
СОАС — синдром обструктивного апное сну.
СТГ — соматотропний гормон.
СХЛ — синдром Хіппеля — Ліндау.
ТТГ — тиреотропний гормон.
УЗ — ультразвук.
ФМД — фібром’язова дисплазія.
ХХН — хронічна хвороба нирок.
ЦОГ — циклооксигеназа.
ЧМТ — черепно–мозкова травма.
ШКФ — швидкість клубочкової фільтрації.