За останні 20 років була розроблена й ліцензована значна кількість протинападових препаратів (ПНП). Однак близько 30 % пацієнтів мають фармакорезистентну епілепсію (ФРЕ), несприйнятливу до звичайних фармакологічних методів лікування. Міжнародна ліга боротьби з епілепсією (ILAE) визначає фармакорезистентну епілепсію як таку, при якій неможливо досягти стійкої ремісії нападів при застосуванні двох препаратів, які добре переносяться, правильно добрані й призначені в адекватних дозах (у монотерапії або в комбінації).
Виходячи з цього визначення, в обсерваційному дослідженні за участю госпіталізованих пацієнтів з уперше діагностованою епілепсією проаналізовано реакцію на ПНП і показано, що перша і друга схеми лікування були успішними в 49,5 і 36 % випадків відповідно. Усі види ПНП-терапії після неефективності перших двох препаратів мали значно нижчий показник успіху (від 12,5 до 22,2 %). Отже, після невдачі другої ПНП-терапії шанси контролювати напади різко знижуються. У зв’язку з цим деякі клініцисти уникають пробувати інші фармакологічні методи лікування в пацієнтів, яким з більш високим відсотком успіху може допомогти хірургічне лікування.
Слід зазначити, що концепція фармакорезистентності враховує не тільки напади, які важко піддаються лікуванню, але й основний патогенез, відповідальний за структурні й нейробіохімічні зміни, що спричиняють когнітивні та нейропсихіатричні розлади, а також психосоціальну дисфункцію. Через значну мінливість фенотипів пацієнтів з ФРЕ і відсутність єдиного визначення ефективності ПНП у досягненні контролю нападів порівняння клінічних випробувань і визначення практичних рекомендацій залишається складним завданням.
Згідно з рекомендаціями ILAE протинападова терапія вважається ефективною, якщо період без нападів становить 12 місяців або принаймні втричі перевищує найдовший інтервал між нападами після початку лікування. Фармакологічна терапія повинна бути підібрана відповідно до епілептичного синдрому, типу нападів і має призначатися протягом як мінімум 6 місяців в адекватному дозуванні. Оптимальний діапазон ефективних доз і частота їх введення залежать від індивідуальної реакції пацієнта, супутніх захворювань, переносимості ліків і їх побічних ефектів.
Отже, пацієнти з ФРЕ не є групою з одним і тим же захворюванням. Вони мають різну клінічну і нейробіологічну картину, що вимагає комплексного підходу до низки питань щодо їх лікування.
Тягар фармакорезистентної епілепсії
Епідеміологічний систематичний огляд L. Kalilani et al. (2018) дозволив встановити, що загальна частка захворюваності на ФРЕ становить 0,06–0,51 %, а її поширеність — 0,11–0,58 %, що узгоджується з результатами інших дослідницьких робіт. Частка захворюваності на ФРЕ у дітей — 0,15 % (95% ДІ: 0,11–0,19), у дорослих — 0,34 % (95% ДІ: 0,06–0,62).
Поширеною причиною неефективності лікування епілепсії є недотримання режиму терапії і неправильний вибір ПНП через помилковий діагноз типу епілептичних нападів. Для забезпечення розуміння проблеми й дотримання запропонованого режиму прийому ліків обов’язковим є створення довірчих відносин з пацієнтами та їх сім’ями. Крім того, у будь-якого пацієнта з важковиліковними нападами епілепсії слід виключити умовну (несправжню) фармакорезистентність. Її причинами можуть бути неправильна діагностика типу нападу епілепсії, призначення неадекватних доз препаратів, неправильна оцінка відповіді на лікування, гіпердіагностика побічних ефектів.
Пацієнти, ідентифіковані як фармакорезистентні, можуть мати фази тривалої повної ремісії, що чергуються з рецидивуючим перебігом. Однак у досліджуваній когорті дорослих з ФРЕ серед пацієнтів з 12-місячним періодом ремісії нападів ризик рецидиву залишався високим — 71,2 % через 5 років. Тому при обговоренні ймовірності стабільної ремісії розумно проявляти обережність.
Примітно, що в людей з ФРЕ ризик раптової смерті у 2–10 разів вище, ніж у загальній популяції. При ФРЕ вона визначається як раптова, несподівана смерть людини, яка страждає від епілепсії і є здоровою за іншими показниками і в якої при проведенні розтину не виявлені інші причини смерті. Ризик раптової смерті особливо великий у пацієнтів з високою частотою судомних нападів і довготривалою епілепсією. У зв’язку з цим єдиним способом запобігання цьому ускладненню є оптимізація контролю над судомними нападами.
До інших причин смерті в пацієнтів з ФРЕ відносять нещасні випадки внаслідок епілептичних нападів, церебральні новоутворення й нейродегенеративні захворювання, які лежать в основі структурних епілепсій. Порівняно із загальною популяцією в дорослих пацієнтів значно частіше виявляються депресія і тривога, у дітей — синдром дефіциту уваги з гіперактивністю, розлади аутистичного спектра і поведінкові проблеми. Ці захворювання негативно впливають на довгострокове психосоціальне функціонування, що обумовлено погіршенням мовленнєвих і соціальних навичок.
Ступінь інвалідності широко варіює залежно від структурної або генетичної етіології епілепсії, яка призвела до нападів або негативного ефекту терапії ПНП. При синдромі дефіциту транспортера глюкози I типу (GLUT1) можуть бути корисними таргетна терапія і кетогенна дієта, які, впливаючи на причину захворювання, здатні зменшити напади й поліпшити когнітивні функції. При епілептичній енцефалопатії своєчасний початок ефективної терапії ПНП може покращити когнітивні й нейроповедінкові симптоми. Однак, незважаючи на адекватну терапію, симптоми супутніх захворювань можуть зберігатися, що, імовірно, обумовлено синаптичною реорганізацією або порушенням нейрогенезу, що створює додаткову проблему їх лікування у пацієнтів із ФРЕ.
У людей з епілепсією також нерідко зустрічаються мігрень, серцево-судинні захворювання, астма, остеоартрит і гастроезофагеальний рефлюкс. Виявлені механізми асоціації варіюють від випадкових, причинних, результуючих, двоспрямованих до загальних факторів ризику. Наприклад, епілепсія може бути викликана цереброваскулярним захворюванням або перинатальним інсультом, тоді як аспіраційна пневмонія, перелом, пов’язаний із судомним нападом, можуть розглядатися як результуюча коморбідність епілепсії. Іноді ці захворювання можуть розглядатися як фактори ризику або як одне генетичне захворювання, відповідальне за обидва стани.
Прогнозовані фактори ризику
Виявлення пацієнтів, які мають більш високий ризик розвитку фармакорезистентної епілепсії, було зроблено в декількох дослідженнях. Однак їх результати не були вірогідними, що обумовлено, з одного боку, відсутністю загального визначення фармакорезистентності в контексті віддаленого прогнозу, а з іншого — –неоднорідністю досліджуваної популяції. Проте діагностика деяких типів епілептичних синдромів у дитячому віці вже означає прогностичну оцінку фармакорезистентності. Так, синдром Леннокса — Гасто, рання дитяча епілептична енцефалопатія, синдром Драве, енцефаліт Расмуссена є фармакорезистентними, що пояснюється вродженим нейробіологічним патерном, який лежить в основі цих типів епілепсії.
ФРЕ асоційовані з такими клінічними факторами, як поява епілепсії в ранньому віці (< 1 року), аномалії мозку, виявлені за допомогою методів нейровізуалізації, нервово-психічні розлади, розумова відсталість, наявність в анамнезі тривалих фебрильних судом або епілептичного статусу, специфічні зміни біоелектричної активності головного мозку на електроенцефалограмі.
Дебют епілепсії в неонатальному періоді пов’язаний з більш високим ризиком розвитку фармакорезистентності, ніж її поява в більш пізньому віці. Ідіопатична епілепсія має нижчий ризик фармакорезистентності порівняно з епілепсією, в основі якої лежать структурні аномалії — кортикальна дисплазія, мезіальний скроневий склероз, туберозний склероз або судинні ураження. Припускається, що напади фокальної епілепсії порівняно з генералізованими пов’язані з більшим ризиком фармакорезистентності.
За даними Chen et al. (2018), прогностичними факторами при ФРЕ є кількість нападів на рік до початку лікування, попередній анамнез зловживання наркотиками і сімейний анамнез епілепсії в родичів першого ступеня. У цілому в когорті пацієнтів з уперше діагностованою епілепсією всіх типів під час лікування першим ПНП нападів позбулися понад 50 %, під час лікування другим і третім ПНП — близько 15 %. Тільки 3 % випадків контролювалися лікуванням двома препаратами.
Для визначення довгострокового результату в пацієнтів з епілепсією прогностичне значення має повне припинення нападів через 6 місяців після початку лікування. Згідно з апостеріорним аналізом пацієнтів, які отримували лікування з приводу фокальної епілепсії, у пацієнтів, у яких не було нападів через 6 місяців, імовірність їх відсутності через 12 місяців становила 90 %.
Патогенез
Механізми, що лежать в основі ФРЕ, повністю не відомі. Імовірно, що патогенез стійкості до лікарських препаратів є варіабельним і багатофакторним процесом за участю декількох механізмів, що діють одночасно в даного пацієнта. Передбачувані механізми лікарської стійкості також можуть мати змішаний характер за участю чинників, пов’язаних із захворюванням, прийомом лікарських засобів і генетичною детермінацією.
Згідно з гіпотезою транспортерів підвищена експресія білків-транспортерів, що забезпечують розподіл лікарських засобів у речовині головного мозку, знижує ефективність ПНП незалежно від мішені їх дії. АТФ-залежний Р-глікопротеїн є продуктом людського гена множинної лікарської стійкості 1 (MDR1; ABCB1), який унаслідок своєї широкої субстратної специфічності відіграє важливу роль в обмеженні надходження в мозок різних ліків. Р-глікопротеїн розташовується в ендотеліальних клітинах капілярів головного мозку, які беруть участь в утворенні гематоенцефалічного бар’єра (ГЕБ). Разом з іншими білками, відповідальними за лікарську стійкість, Р-глікопротеїн захищає мозок від токсичного пошкодження ліпофільними ксенобіотиками, які можуть проникати через ГЕБ шляхом пасивної дифузії. Цим механізмом у низці випадків обумовлена неефективність терапії пухлин та інфекцій головного мозку.
Деякі ПНП мають хімічну структуру, подібну до субстратів P-глікопротеїну, що обмежує їх проникнення через ГЕБ і сприяє виникненню множинної лікарської стійкості. Разом з тим дані, що підтверджують гіперекспресію Р-глікопротеїну в епілептогенних вогнищах головного мозку, суперечливі, як і роль інших білків, що беруть участь у розвитку фармакорезистентності. Здається, гіперекспресія білків-транспортерів обмежується лише епілептичним вогнищем, не впливаючи на сусідні нормальні тканини, що пояснює значно меншу кількість побічних ефектів у пацієнтів з ФРЕ, ніж у пацієнтів, які реагують на лікування.
Як свідчать експериментальні дослідження, у щурів, які не відповіли на лікування ПНП, виявляється більш висока експресія Р-глікопротеїну. При його інгібуванні тариквідаром резистентність до ПНП знижується.
Отже, гіпотеза транспортерів все ще залишається спірним питанням, прояснити яке дозволять результати нових досліджень з оцінкою гіперекспресії транспортерів у ГЕБ.
Фармакокінетична гіпотеза ФРЕ припускає, що гіперекспресія білків-транспортерів локалізована в периферичних органах, таких як печінка, кишечник і нирки. У зв’язку з цим відзначається зниження рівня ПНП у плазмі крові, а його кількість стає недостатньою для проникнення через гематоенцефалічний бар’єр. Однак дослідження, проведені на тваринах, не підтверджують цю гіпотезу.
Існує також гіпотеза мішені, яка постулює, що набуті, індуковані епілепсією зміни структури або функції молекул — мішеней ПНП призводять до зменшення відповіді на лікування. У першу чергу дана теорія заснована на дослідженнях дії карбамазепіну й фенітоїну на потенціалзалежні натрієві канали в нейронах гіпокампа в пацієнтів зі скроневою фокальною епілепсією. Їх блокування було повністю втрачено в пацієнтів з лікарською стійкістю до карбамазепіну і фенітоїну порівняно з пацієнтами, які не мають резистентності. Досі залишається неясним, чи може втрата чутливості натрієвих каналів у пацієнтів з резистентністю до даних препаратів поширюватися на інші ПНП з аналогічними механізмами дії.
Відповідно до гіпотези мішені були вивчені інші точки застосування, наприклад рецептори ГАМК. Повідомлялося про зміну їх чутливості в експериментальній моделі ФРЕ у тварин, однак підтвердження в клінічних дослідженнях вони не отримали.
Крім того, епілепсія може спричинити структурні зміни — нейродегенерацію, розростання аксонів, синаптичну реорганізацію, нейрогенез і гліоз. Ці зміни викликають формування аномальної нейронної мережі, яка може призвести до резистентності ПНП. Так, при скроневій епілепсії причинну роль у виникненні фармакорезистентності відіграє склероз гіпокампа, тоді як його резекція призводить до зникнення лікарської резистентності. Враховуючи, що аномалія нейронної мережі не завжди супроводжується рефрактерністю, можна припустити, що, імовірно, її розвиток зумовлений кількома причинними факторами.
Гіпотеза внутрішньо властивої тяжкості перебігу, навпаки, розглядає фармакорезистентність як невід’ємну властивість епілепсії, пов’язану з тяжкістю захворювання. Зручним орієнтиром для оцінки тяжкості є частота нападів до початку лікування. Дослідники виходять з того, що епілептичні напади можливі в будь-якої людини, при цьому серед хворих з епілепсією в одних вони виникають щодня, а в інших — раз на рік або рідше. Даний феномен може пояснюватися відмінністю в сприйнятливості нейронів головного мозку до певного фізіологічного стимулу, що індукує епілептичну активність. Висока частота пароксизмів і, відповідно, тяжкість епілепсії вказує на низький поріг чутливості нейронів, а фармакорезистентність є лише наслідком цієї нестабільності.
Генетична гіпотеза припускає, що низька чутливість до ПНП пов’язана з однонуклеотидним поліморфізмом у генах SCN1A, SCN2A і SCN3A. Показана роль мутацій у цих генах у патогенезі деяких форм епілепсії (наприклад, синдрому Драве), і висловлюється припущення про те, що вони можуть мати значення і при ФРЕ.
Нейрозапалення і дисфункція ГЕБ розглядаються як потенційні механізми стимуляції та підтримки епілептичної активності. Підвищена проникність ГЕБ присутня як в експериментальних моделях, так і в клінічних умовах і зазвичай пов’язана із супутньою ней-розапальною реакцією в тих же ділянках тканини. У всіх цих умовах у судинах і астроцитах головного мозку відбувається індукція P-глікопротеїну.
Отже, нейрозапалення і дисфункція ГЕБ є ознаками епілептогенної зони при різних формах ФРЕ. У доклінічних дослідженнях на тваринних моделях було доведено, що нейрозапалення призводить до гіперзбудливості нейронів. Мікроглія та астроцити відіграють вирішальну роль як в індукції, так і в підтримці запальної реакції на епілептогенні пошкодження або напади. Іншими учасниками є нейрони, клітинні компоненти ГЕБ і лейкоцити.
Крім того, були виявлені специфічні запальні молекули та шляхи, що впливають на результати в різних експериментальних моделях епілепсії. Усе більше доказів свідчить про те, що нейрозапалення, зміни проникності ГЕБ, астроцитарна дисфункція призводять до епілептогенезу, прогресування епілепсії та її резистентності до ПНП.
Антитіло-опосередковані енцефалопатії являють собою запальні захворювання головного мозку, які все частіше визнаються причиною нападів та епілептичного статусу. Вони стійкі до звичайних ПНП, але можуть ефективно усуватися імунотерапією. Рання діагностика цих станів приводить до своєчасного лікування імуномодулюючими агентами, тому в більшості випадків симптоматичне лікування ПНП може бути припинено після гострої фази захворювання. При цьому ризик розвитку хронічної епілепсії відносно низький (10–15 %) і може варіювати залежно від антигену-мішені та своєчасної імунотерапії.
Медикаментозна терапія фармакорезистентної епілепсії: монотерапія порівняно з раціональною політерапією в епоху нових ПНП
Незважаючи на значний прогрес у лікуванні епілепсії та появу нових ПНП, терапія ФРЕ все ще залишається складним завданням. Основою її лікування є медикаментозна терапія, підбір якої здійснюється з урахуванням співвідношення ризику/користі, ефективності, переносимості й комплаєнсу.
Для лікування пацієнтів з нещодавно діагностованою епілепсією слід розглянути монотерапію. Після невдачі принаймні двох випробуваних режимів лікування епілепсії слід розглянути політерапію. Відбір пацієнтів з ФРЕ на політерапію, пошук і вивчення нових ПНП є найбільш важливими завданнями для лікаря.
Застосування ПНП першого покоління мало суттєві обмеження, пов’язані з фармакокінетичними властивостями препаратів (сильні індуктори або інгібітори печінкових ферментів) та високою частотою небажаних явищ. Було проведено тільки одне рандомізоване контрольоване дослідження (РКД), у якому на початку лікування пацієнтам з генералізованими тоніко-клонічними і/або фокальними нападами призначали карбамазепін або комбіновану терапію карбамазепіном і вальпроатом. Результати дослідження показали переваги комбінованої терапії в зменшенні частоти нападів.
Перед виходом на фармацевтичний ринок нові ПНП проходять доклінічні й ретельно сплановані рандомізовані клінічні дослідження за участю пацієнтів, які приймають від одного до трьох ПНП. Результати цих досліджень показали перевагу нових ПНП як додаткової терапії порівняно з плацебо. Були отримані докази їх кращого фармакологічного профілю, включно з лінійною фармакокінетикою, менший потенціал для лікарських взаємодій і різні механізми дії, які можуть поєднуватися один з одним у політерапії. Після багатьох років використання нові ПНП були вивчені в порівняльних РКД як моно- і політерапія. Деякі з них у даний час показані як препарати першої лінії при фокальних нападах або певних типах епілепсії.
Метою політерапії у фармакорезистентних пацієнтів є оптимальний підбір ПНП, які не тільки посилюють ефективність один одного, але й мінімізують побічні ефекти. Клінічні дослідження з підбором найкращих комбінацій ПНП на людях обмежені, отже, вибір другого або третього препарату в раціональній політерапії проводитися емпірично або на підставі результатів експериментальних досліджень. Експериментальні моделі на тваринах дозволили оцінити фармакодинамічні взаємодії препаратів з різними механізмами дії при використанні комбінованої терапії. Бажаними при її застосуванні повинні бути протинападовий супраадитивний (синергічний) ефект і нейротоксичний інфраадитивний ефект (нейротоксичний антагонізм).
У клінічних випробуваннях було показано, що більша частота побічних явищ і нижча ефективність супроводжують комбінації ПНП з однаковими ефектами блокування натрієвих каналів порівняно з комбінованою терапією блокатором натрієвих каналів і препаратом з іншим механізмом дії, наприклад ГАМКергічним препаратом. У великому популяційному дослідженні, у якому брали участь пацієнти з фокальними нападами епілепсії, було виявлено, що поєднання ПНП з різними механізмами дії демонструє кращі результати щодо тривалості лікування, ризику госпіталізації та госпіталізації до відділень невідкладної допомоги.
Кращу синергічну взаємодію мають вальпроат і ламотриджин. Про це повідомлялося в кількох дослідженнях, які показали більш високий рівень відповіді при застосуванні вальпроату й ламотриджину як додаткової терапії порівняно з додаванням ламотриджину до карбамазепіну або фенітоїну.
У дітей з абсансами, які важко піддаються лікуванню, поєднання вальпроату з етосуксимідом було більш ефективним, ніж монотерапія даними препаратами. Іншими схемами комбінованої терапії, вивченими в когорті пацієнтів з фокальними нападами, є ламотриджин + леветирацетам і лакосамід + леветирацетам. Синергічний ефект комбінації цих ПНП може бути результатом об’єднання різних механізмів дії. Дійсно, лакосамід посилює інактивацію залежних від потенціалу натрієвих каналів, тоді як леветирацетам шляхом зв’язування білка синаптичних везикул SV2A в мозку забезпечує вивільнення синаптичних нейромедіаторів. Комбіновані режими ламотриджин + топірамат і вальпроат + леветирацетам у дорослих пацієнтів також можуть бути корисними для зменшення нападів, хоча вони мають низький рівень доказів.
Є дані про успішне використання комбінації клобазаму* або вальпроату із стирипентолом як додаткової терапії в дітей і підлітків із синдромом Драве. Для додаткової терапії стирипентол схвалений у Європі (2007), Японії (2012), Канаді (2012) і США (2018).
Два контрольовані випробування, проведені у 2000 і 2002 роках за участю молодих пацієнтів із синдромом Драве, які отримували лікування вальпроатом або клобазамом*, показали більш високу частоту відповіді в пацієнтів при його доповненні стирипентолом* порівняно з плацебо. Ці результати були підтверджені подальшими спостережними ретроспективними і проспективними довгостроковими дослідженнями, які показали перевагу додавання стирипентолу* щодо контролю нападів, зменшення їх тривалості, кількості епізодів епілептичного статусу і госпіталізацій. Протинападові ефекти стирипентолу*, що спостерігаються при синдромі Драве, можуть бути результатом двох різних механізмів. Один з них, фармакокінетичний, забезпечує збільшення активних метаболітів клобазаму*, опосередковане стирипентолом*, інший стосується механізму дії стирипентолу*, що заснований на посиленні передачі гамма-аміномасляної кислоти через постсинаптичні рецептори ГАМК у місці дії, відмінному від такого в бензодіазепінів.
Нарешті, інші нові ПНП, такі як еслікарбазепіну ацетат* габапентин і зонісамід, виявилися ефективними як додаткові препарати при лікуванні фармакорезистентної епілепсії. Проте навіть з огляду на зростаючий інтерес до нових ПНП і рекомендації щодо їх використання для лікування ФРЕ у порівняльних дослідженнях жоден не довів своєї переваги в ефективності зменшення нападів над звичайними ПНП.
Іншими проблемами, пов’язаними з використанням раціональної політерапії, є різні небажані явища і наслідки міжлікарських взаємодій. Як згадувалося раніше, ПНП першого покоління мають багато взаємодій з іншими ліками, у тому числі з ПНП, що обумовлено їх дією як індукторів або інгібіторів цитохрому Р450. Так, при призначенні вальпроату й ламотриджину необхідно враховувати, що вальпроат діє як потужний інгібітор ферментативного метаболізму, знижуючи кліренс ламотриджину і підвищуючи рівень його в крові, що збільшує імовірність розвитку ламотриджин-індукованої гіперчутливості, постурального й рухового тремору. Це обумовлює необхідність повільного титрування ламотриджину, починаючи з більш низьких доз і підвищуючи їх за необхідності. Карбамазепін, фенобарбітал і фенітоїн є індукторами цитохрому Р450, унаслідок чого можуть знижувати рівні антикоагулянтів, оральних контрацептивів та імунодепресантів.
ПНП першого покоління мають спектр побічних явищ, які варіюють від гепатотоксичності та енцефалопатії, пов’язаних з прийомом вальпроату, до пригнічення функцій кісткового мозку, викликаного карбамазепіном, фенітоїном, фенобарбіталом. На відміну від них нові ПНП відзначаються меншою фармакокінетичною взаємодією (більшість з них є слабкими індукторами або інгібіторами ферментів) і мають кращі профілі переносимості, отже, вони є оптимальними кандидатами для комбінованої терапії.
Леветирацетам має найменшу кількість фармакологічних взаємодій і разом з топіраматом і зонісамідом широко використовується в політерапії. Деякі нові ПНП мають специфічні протипоказання: лакосамід, руфінамід* і ретигабін* не рекомендуються до застосування в пацієнтів із синдромом подовженого інтервалу QT, а топірамат, зонісамід і леветирацетам через підвищений ризик розвитку тривоги, депресії, психозу — у пацієнтів із супутніми психоневрологічними захворюваннями.
Слід зазначити, що використання політерапії не означає обов’язкового збільшення небажаних явищ (НЯ). Згідно з результатами досліджень, ризик НЯ був однаковим у пацієнтів, які отримували монотерапію і політерапію. При цьому пацієнти, які належали до другої групи, були здатні переносити більш високе загальне лікарське навантаження порівняно з пацієнтами, які отримували монотерапію. Автори припустили, що виникнення НЯ при політерапії пов’язано не з кількістю використовуваних препаратів, а з типом і дозуванням обраних ПНП, а також з індивідуальною сприйнятливістю до них.
Отже, у рамках раціональної терапії ФРЕ при виборі оптимальної схеми необхідно враховувати безліч змінних, що стосуються не тільки фармакодинамічних і фармакокінетичних аспектів препаратів, але і факторів, пов’язаних з пацієнтом, таких як тип нападів, вік, дотримання режиму лікування, супутні захворювання і ліки.
Протинападові препарати при рефрактерних епілептичних синдромах
Ідіопатичні епілептичні синдроми включають чотири форми генералізованих епілепсій: дитяча і юнацька абсансна, юнацька міоклонічна епілепсія та епілепсія з ізольованими тоніко-клонічними нападами. Як правило, вони трапляються в дітей і підлітків, мають відоме або передбачуване генетичне походження і впізнавану електроенцефалографічну й клінічну картину. У більшості пацієнтів з ідіопатичними епілептичними синдромами спостерігається спонтанна ремісія або зникнення нападів на тлі лікування. Однак у 20–30 %, незважаючи на терапію ПНП першої та другої лінії, напади можуть зберігатися, що свідчить про фармакорезистентність епілептичного синдрому.
Для лікування фармакорезистентного синдрому дитячої абсансної епілепсії, який не реагує на монотерапію етосуксимідом*, вальпроатом і ламотриджином, може бути використана комбінація вальпроату й ламотриджину. Ця комбінація має синергічну дію і може бути більш ефективною, ніж лікування кожним препаратом окремо. Комбінація вальпроату й етосуксиміду* також ефективніша в пацієнтів з фармакорезистентним синдромом дитячої абсансної епілепсії при несприйнятливості до монотерапії етосуксимідом* або вальпроатом. При невдачі цих варіантів лікування можна застосовувати клобазам*, зонісамід, топірамат. Фенітоїн, карбамазепін і барбітурати неефективні й протипоказані при абсансах.
Енцефалопатії розвитку й епілептичні енцефалопатії при синдромі Леннокса — Гасто, синдромі Драве, ранні дитячі епілептичні енцефалопатії майже завжди вимагають комбінованої терапії. У контексті онтогенетичних та епілептичних енцефалопатій синдром Драве є прототипом фармакорезистентного епілептичного синдрому. Це генетично детермінована патологія, що виникає внаслідок мутації локусу SCN1A, який кодує потенціал-керовані натрієві канали, що призводить до порушення фізіологічних процесів реполяризації і деполяризації в нейронах і, як наслідок, до патологічної активності центральної нервової системи. Як лікування 1-ї лінії при синдромі Драве призначається вальпроат або клобазам* у поєднанні зі стирипентолом*. Використання топірамату може розглядатися як альтернатива стирипентолу* при додаванні до клобазаму* або вальпроату.
Крім того, клінічні випробування продемонстрували ефективний контроль над епілептичними нападами нових фармакологічних методів лікування синдрому Драве — канабідіолу*, фенфлураміну*. Канабідіол* може бути використаний для додавання до інших ПНП як терапія другої лінії. Є багатообіцяючі клінічні докази використання фенфлураміну* в низьких дозах при синдромі Драве. Однак його ефективність і безпека ще не підтверджені.
Синдром Леннокса — Гасто є однією з найтяжчих дитячих епілептичних енцефалопатій, на нього припадає 1–10 % усіх дитячих епілепсій. Пік захворюваності — 3–5 років. Синдром Леннокса — Гасто має відносно гетерогенну етіологію (генетичну, структурну, метаболічну або невідому), специфічну картину множинних типів нападів (тонічні, атонічні, дроп-атаки), порушення ЕЕГ і типову рефрактерність до лікування з короткими періодами ремісії. У даний час для цих пацієнтів відсутні РКД монотерапії і прямого порівняння додаткових ПНП. Вальпроат досі вважається терапією першої лінії для лікування нещодавно діагностованого синдрому Леннокса — Гасто. У разі невдачі вальпроату кращим вибором є ламотриджин. Він має найвищу ефективність у контролі дроп-атак, проте протипоказаний при міоклонічних судомах.
Показано, що руфінамід* і клобазам* ефективні в комбінованій терапії з добрим рівнем доказів. Зонісамід, леветирацетам, перампанель* також можуть мати ефективність при цьому синдромі. Канабідіол* треба призначати в тих випадках, коли інші методи лікування виявилися неефективними.
Практичний підхід до ведення пацієнтів з фармакорезистентною епілепсією
Для лікування пацієнтів з нещодавно діагностованою епілепсією показана монотерапія. Її використання дозволяє досягти тривалого контролю над нападами (> 12 місяців) у 50 % пацієнтів. На думку деяких експертів, політерапію слід розглядати лише після неефективності принаймні двох-трьох ПНП. Проте нещодавно проведені дослідження свідчать, що при неефективності початкової монотерапії як альтернатива другому препарату має бути розглянута дуотерапія. Це дозволяє досягти ремісії нападів ще в 15–20 % пацієнтів.
Пацієнти, які не відповіли на друге лікування, чи то монотерапію, чи то дуотерапію, задовольняють критеріям ILAE щодо фармакорезистентної епілепсії і повин-ні бути направлені в спеціалізовані центри епілепсії для подальшого спостереження й підбору лікування. Їм проводиться пролонгована відео-ЕЕГ з нейровізуалізацією, що дозволяє встановити правильний діагноз типу епілепсії і виключити причини псевдофармакорезистентності. Пацієнти, які можуть отримати користь від хірургічного лікування, наприклад, при фокальних епілептогенних ураженнях, мають більш високу ймовірність ремісії нападів (близько 60–80 %) при своєчасному направленні в спеціалізовані центри. Якщо ураження неможливо усунути хірургічним шляхом без подальших неврологічних ускладнень, може знадобитися підбір другої та третьої терапії.
Отже, вибір кандидатів на політерапію є важливим кроком щодо збільшення можливості контролю нападів. Однак слід уникати додавання четвертого препарату, оскільки використання понад трьох препаратів підвищує імовірність НЯ порівняно з незначним поліпшенням контролю за нападами. Якщо 5-те або 6-те лікарське випробування з дуо -, потрійною або квадротерапією не вдалося, слід оцінити інші альтернативні методи лікування — стимуляцію блукаючого нерва або кетогенну дієту. Якщо пацієнт вже отримував лікування ПНП, украй важливо оцінити дози введення, ефективність і спектр небажаних явищ раніше призначених препаратів.
Нові протинападові препарати для подолання фармакорезистентної епілепсії
За останні десятиліття на фармацевтичному ринку з’явилося багато нових ПНП. Однак, незважаючи на це, 30 % пацієнтів з епілепсією не реагують на медикаментозну терапію і гостро потребують нових варіантів лікування для контролю нападів і поліпшення якості життя.
У 2004 р. Американська академія неврології (AAN) та Американська асоціація епілепсії (AES) опублікували рекомендації щодо 8 протисудомних препаратів другого покоління (табл. 1).
Уже після їх публікації Управління з контролю за продуктами та ліками (FDA) схвалило 6 нових ПНП і 2 ПНП другого покоління (табл. 2).
Як додаткова терапія для зниження частоти нападів у дорослих пацієнтів з фармакорезистентною фокальною епілепсією були досліджені такі препарати: прегабалін, лакосамід, руфінамід*, езогабін*, вігабатрин, клобазам*, перампанель*, еслікарбазепін*, окскарбазепін пролонгованої дії, топірамат пролонгованої дії.
На основі результатів досліджень AAN та AES рекомендують застосування прегабаліну з негайним вивільненням і перампанелю* для лікування фармакорезистентної фокальної епілепсії в дорослих пацієнтів; вігабатрину — при фармакорезистентній фокальній епілепсії як 2-гу й наступні лінії терапії. Езогабін* може зменшувати частоту нападів у цій популяції, але його використання пов’язане із серйозним ризиком знебарвлення шкіри й сітківки (рівень В).
Нещодавно також схвалений бриварацетам* як додаткова терапія в пацієнтів з фокальними нападами, з переходом у двобічні тоніко-клонічні напади або без нього, а також з первинно-генералізованими тоніко-клонічними нападами. Подібно до леветирацетаму бриварацетам* має високу й селективну активність щодо білка синаптичних везикул SV2A і лінійну фармакокінетику. У дорослих пацієнтів і дітей з резистентною до ліків фокальною епілепсією було доведено, що призначення бриварацетаму* ефективно зменшує частоту нападів і добре переноситься. Однак, щоб зробити остаточні висновки про ефективність препарату в учасників, які не отримували леветирацетам, а також оцінити його довгостроковий профіль безпеки, необхідне проведення подальших досліджень.
Керівництво Американської академії неврології також повідомляє про можливість використання монотерапії ламотриджином, окскарбазепіном і топіраматом при фокальній ФРЕ. Пізніше проводилися дослідження з леветирацетамом з пролонгованим вивільненням у дозі 1000 і 2000 мг/добу (1 дослідження), прегабаліном 150 і 600 мг/добу (1 дослідження), лакосамідом 300 і 400 мг/добу (1 дослідження) та еслікарбазепіном* 1200 і 1600 мг/добу (2 дослідження). Результати цих досліджень продемонстрували, що серед нових ПНП тільки еслікарбазепін* є ефективним препаратом для використання в монотерапії вогнищевої ФРЕ.
У керівництві AAN габапентин, ламотриджин, окскарбазепін і топірамат рекомендовані як додаткова терапія при лікуванні фармакорезистентної фокальної епілепсії в дітей. Відтоді було опубліковано результати 4 досліджень: 2 — для леветирацетаму, 1 — для окскарбазепіну та 1 — для зонісаміду. Після їх оцінки експерти рекомендували використання зонісаміду при фармакорезистентній фокальній епілепсії в дітей 6–17 років; окскарбазепіну — при фокальній ФРЕ в дітей віком від 1 місяця до 4 років.
У керівництві AAN топірамат рекомендується при фармакорезистентних генералізованих тоніко-клонічних нападах (ФР ГТКН) у дорослих і дітей для додаткової терапії. Пізніше були проведені й опубліковані 3 дослідження із застосування ламотриджину і 2 — з використання леветирацетаму.
Ламотриджин для лікування ФР ГТКН застосовували в декількох дозах (залежно від віку і типу використовуваного додаткового ПНП) і порівнювали з плацебо в 1 дослідженні класу II за участю пацієнтів віком 2–55 років. Середнє відсоткове зниження частоти ГТКН за 12 тижнів і показники відповіді були значно вищими в пацієнтів, рандомізованих у групу ламотриджину. Найпоширеніші НЯ, пов’язані з його прийомом, включали запаморочення, сонливість і нудоту.
Друге дослідження класу II продемонструвало значні відмінності в середньому відсотковому зниженні порівняно з базовим рівнем нападів ГТКН на користь ламотриджину як у періоди ескалації, так і в періоди підтримуючої терапії. НЯ, що призвели до відміни препарату, були рідкісні, про висипання не повідомлялося.
Подібні результати були отримані в дослідженні класу I, у якому брали участь підлітки й дорослі, які приймали ламотриджин з пролонгованим вивільненням (LTG-XR). У тих, хто приймав ламотриджин пролонгованого вивільнення, спостерігалося значно більше середнє відсоткове зниження щотижневої частоти нападів ГТКН і більш висока частота відповіді на терапію. Нудота, блювання й диплопія були найбільш частими НЯ, пов’язаними з прийомом препарату.
Було проведено 2 дослідження класу I, у яких порівнювали леветирацетам у дозі 3000 мг/день і плацебо для лікування ФР ГТКН. Одне дослідження включало пацієнтів віком від 4 до 65 років. У даному дослідженні леветирацетам показав більш високу частоту відповіді і частоту відсутності ГТКН. У другому дослідженні оцінювали підлітків і дорослих з ювенільною міоклонічною епілепсією або ювенільною абсансною епілепсією. Було виявлено, що в більшої кількості пацієнтів, які приймали леветирацетам, відмічалося 50% зниження кількості днів на тиждень з міоклонічними нападами.
На підставі цього експерти AAN дійшли висновку, що ламотриджин як з негайним, так і з пролонгованим вивільненням ефективний як додаткова терапія ФР ГТКН (1 дослідження класу І для LTG-XR; 2 дослідження класу II для ламотриджину з негайним вивільненням) (рівень B). Також слід розглянути можливість використання леветирацетаму при ФР ГТКН, ювенільній міоклонічній епілепсії та ювенільних абсансних нападах (рівень В).
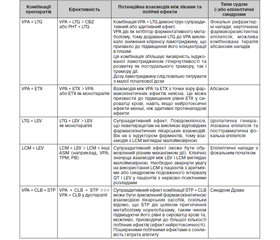
Дані низки досліджень щодо ефективності використання комбінацій деяких протиепілептичних препаратів наведені в табл. 3 (Fattorusso А. et al., 2021). Комбінація вальпроату з ламотриджином широко оцінювалась у кількох дослідженнях і продемонструвала найкращу ефективність.
У рекомендаціях 2004 року фелбамат*, ламотриджин і топірамат були визнані ефективними при лікуванні синдрому Леннокса — Гасто. Відтоді були опубліковані дослідження клобазаму* і руфінаміду*.
У двох дослідженнях класу I порівнювали руфінамід* у дозі до 45 мг/кг/добу з плацебо в дітей і молодих людей із синдромом Леннокса — Гасто. Пацієнти, які були рандомізовані до групи руфінаміду*, мали значно більший відсоток зниження загальної частоти нападів і падінь, а також зменшення тоніко-атонічних нападів порівняно з плацебо. Загальні НЯ включали сонливість, блювання і зниження апетиту.
Ефективність клобазаму* в дозах 0,25; 0,5 і 1 мг/кг порівнювали з плацебо у 2 дослідженнях класу II. Щотижнева частота нападів була значно знижена при застосуванні всіх 3 доз клобазаму* з більш вираженим зниженням в групі, що приймала 1 мг/кг. Найбільш часті НЯ включали сонливість, млявість, седативний ефект, гіперсекрецію слини, запор, агресію, гіпоманію і безсоння. Млявість, атаксія, стомлюваність і агресія були найбільш частими небажаними явищами, що приводили до відміни препарату.
На основі цих досліджень руфінамід* (рівень А) і клобазам* (рівень В) як додаткова терапія рекомендуються для лікування синдрому Леннокса — Гасто.
Серед нових молекул, вивчених в клінічних випробуваннях при ФРЕ, свою ефективність продемонстрували канабідіол* і фенфлурамін*. В одному з перших клінічних випробувань канабідіол* (непсихоактивна сполука, отримана з конопель) був схвалений для використання в комбінованій терапії з клобазамом* у пацієнтів із синдромом Драве і синдромом Леннокса — Гасто в пацієнтів віком від 2 років.
За своєю хімічною структурою і механізмом дії канабідіол* відрізняється від інших ПНП і є першим у новому класі препаратів. Канабідіол* не проявляє психоактивної дії, натомість демонструє виражений протисудомний ефект, впливаючи на антагонізм рецептора 55, пов’язаного з G-білком (GPR55), десенсибілізацію транзиторного рецепторного потенціалу ванілоїдних каналів 1-го типу (TRPV1) і позитивну алостеричну модуляцію ГАМК-рецепторів. В одному рандомізованому подвійному сліпому плацебо-контрольованому дослідженні І фази за участю дітей і підлітків, а також у двох інших РКД ІІІ фази була показана ефективність і безпека канабідіолу* як додаткового препарату в пацієнтів із синдромом Леннокса — Гасто, особливо для контролю дроп-атак. У подальших дослідженнях було виявлено, що додатковий прийом канабідіолу* як у дозі 10 мг/кг/день, так і в дозі 20 мг/кг/день приводить до аналогічного зниження частоти судомних нападів з кращим профілем безпеки для дозування 10 мг. Поширеними небажаними явищами, пов’язаними із застосуванням канабідіолу*, були блювання, стомлюваність, лихоманка, зниження апетиту, сонливість, млявість і діарея. Передбачається, що терапевтичні й побічні ефекти канабідіолу* можуть бути пов’язані з його одночасним прийомом з клобазамом*, оскільки він є інгібітором ферменту цитохрому Р450. При цьому є переконливі докази того, що канабідіол* ефективний як у поєднанні з клобазамом*, так і без нього.
Окрім вивчення канабідіолу* і фенфлураміну*, з’являються багатообіцяючі дослідження щодо використання ценобамату* і падсевонілу*, обговорені в роботі Loscher et al. Відповідно до згаданої вище гіпотези мішені одним з підходів є розробка нових ПНП, орієнтованих на мішень. У зв’язку з цим був створений падсевоніл*, який за механізмом дії схожий на леветирацетам і бриварацетам*, але з більшою спорідненістю до пресинаптичних білків SV2 і більш широкою дією на різні підтипи SV2. У даний час падсевоніл* проходить клінічні випробування ІІІ фази за участю пацієнтів з фокальними нападами і множинною лікарською стійкістю. Ценобамат* — це новий ПНП, нещодавно схвалений для лікування фокальних нападів у дорослих пацієнтів. Його механізм дії заснований на посиленні гальмівних струмів за допомогою модуляції рецепторів GABA і зменшенні збудливого натрієвого струму.
Ще один цікавий підхід в епоху геноміки — «точна медицина». Поява геномних технологій дозволяє виявити генетичний фон епілепсії і змінює класифікацію епілептичних синдромів. Різні типи генних мутацій можуть лежати в основі того самого епілептичного синдрому і відповідати за різну реакцію на ліки. Список генів, що несуть рідкісні патогенні варіанти, швидко зростає. Ці висновки привели до розробки раціональних стратегій лікування, включно з кращим вибором ПНП з існуючих або перепрофільованих препаратів, які раніше не використовувалися для лікування епілепсії. Дійсно, у деяких випадках специфічні метаболічні дефекти можна скорегувати. Наприклад, кетогенна дієта ефективна при дефіциті GLUT1, піридоксин — при піридоксинзалежних епілепсіях. ПНП — блокатори натрієвих каналів протипоказані при синдромі Драве, пов’язаному з дефектом SCN1A, і показані при епілепсії, пов’язаній з мутацією SCN8A або SCN2A. Мемантин використовують для лікування епілептичної енцефалопатії, викликаної мутацією GRIN2A рецепторів глутамату NMDA. Застосування еверолімусу* ефективне при фокальній епілепсії, пов’язаній з туберозним склерозом, використання канабідіолу* і фенфлураміну* — при синдромі Драве.
Альтернативи фармакологічному лікуванню лікарсько-стійкої епілепсії
Кращим альтернативним варіантом ПНП для пацієнтів з рефрактерною епілепсією є хірургічне лікування в тому випадку, коли його проведення видається можливим. Як правило, відтермінування операції зменшує шанси на припинення нападів, тому вкрай важливо виявляти пацієнтів — кандидатів на хірургічне втручання. Показаннями для його проведення є вогнищеві ураження при фокальній епілепсії — склероз гіпокампа і фокальна кортикальна дисплазія, підтверджені за допомогою магнітно-резонансної томографії.
Однак у значної кількості пацієнтів епілептогенна зона розташовується в межах функціональної тканини головного мозку, що унеможливлює проведення хірургічного втручання. Для цієї групи пацієнтів більш прийнятною альтернативою або доповненням до медикаментозної терапії є нейростимуляція. У даний час для лікування ФРЕ доступно кілька методів нейростимуляції: інвазивні, що вимагають хірургічного втручання для імплантації пристроїв, або неінвазивні, без необхідності імплантації постійного пристрою. Деякі пристрої забезпечують безперервну стимуляцію (відкритий цикл), тоді як інші забезпечують стимуляцію виявленої мозкової діяльності (замкнутий цикл). Серед інвазивних методів найбільш вивченим підходом є нейростимуляція блукаючого нерва (VNS), глибока стимуляція мозку (DBS), нейростимуляція пізньої реакції (RNS) і хронічна підпорогова стимуляція кори (CSCS). До неінвазивних належать транскраніальна стимуляція блукаючого нерва (tVNS), стимуляція трійчастого нерва (TNS), транскраніальна магнітна стимуляція (TMS) і транскраніальна стимуляція постійним струмом (tDCS).
Як додаткове лікування ФРЕ у 1997 році Управлінням з контролю за продуктами та ліками була затверджена нейростимуляція блукаючого нерва. Протисудомний ефект VNS обумовлений підвищенням рівня гальмівного нейромедіатора ГАМК і зниженням рівня збудливої амінокислоти аспартату. Крім того, при VNS змінюється функція лімбічної системи, ретикулярної активуючої системи і кортикальних структур (орбіто-фронтальна кора, скроневі частки).
До немедикаментозних методів лікування ФРЕ також належить кетогенна дієта. Вона являє собою строгий дієтичний режим, зазвичай призначений для певної групи дітей з ФРЕ, характеризується високим вмістом жирів і низьким вмістом вуглеводів, що імітує голодування. Через небезпеку негативного впливу на ріст і загальний стан здоров’я кетогенну дієту не використовують довгостроково. Проте при добрій переносимості вона може залишатися допустимим варіантом для пацієнтів з фармакорезистентною епілепсією.
Висновки
Ведення пацієнтів з фармакорезистентною епілепсією все ще залишається серйозною проблемою для лікарів. Неоднорідність клінічних проявів як щодо семіотики, так і щодо перебігу нападів з непостійними і часто непередбачуваними періодами ремісії і рецидивів у пацієнтів з ФРЕ ускладнює порівняння клінічних досліджень і визначення рекомендацій. Відомі в даний час патогенетичні теорії епілепсії, які засновані в основному на доклінічних дослідженнях, не дають однозначного комплексного пояснення фармакорезистентності, а пояснюють лише деякі механізми, що можуть стати мішенню для нових молекул, розроблених у результаті досліджень.
Фармакологічне лікування, незважаючи на неминучі побічні ефекти й міжлікарські взаємодії, залишається першим і основним підходом до досягнення довгострокового контролю над нападами. Після правильної діагностики типу епілепсії і відбору пацієнтів — кандидатів на резекційну хірургію для пацієнтів з ФРЕ необхідний підбір комбінації препаратів з оптимальним режимом їх прийому, адаптований до кожного пацієнта індивідуально. Це вимагає необхідних знань про фармакодинамічний і фармакокінетичний профіль ПНП, їх можливі побічні ефекти й найефективніші фармакологічні комбінації.
У нову еру другого, третього і останнього покоління ПНП раціональна політерапія набула більшої актуальності, що обумовлено розробкою препаратів з різними і потенційно синергічними механізмами дії, а також кращими профілями безпеки й ефективності порівняно з ПНП першого покоління. Тому, перш ніж вважати пацієнта повністю несприйнятливим до медикаментозної терапії, необхідно провести повторну оцінку діагнозу самої епілепсії і, коли це можливо, генетичного фону, критично розглянути попередні препарати, щоб спробувати наступні, більш адекватні терапевтичні схеми.
Альтернативні немедикаментозні підходи, такі як електрична стимуляція та дієтотерапія, також є перспективними, але вони не є вирішальними в довгостроковій перспективі.

/25.jpg)
/26.jpg)