Вступ
Зараз у світі все більш успішно застосовують генні методи дослідження, особливо генетичне секвенування. Разом із цим значно розширились наші знання про генетичні зміни, які відбуваються в усьому геномі людини, що дозволило швидко та ефективно виявляти гени, пов’язані з багатьма захворюваннями. У даній статті ми розберемо кілька клінічних випадків, у яких ключову роль відіграло генетичне секвенування в діагностиці рідкісних неврологічних захворювань, та розглянемо подальшу тактику ведення таких пацієнтів.
Епілепсія може бути наслідком первинних генетичних аномалій або вторинною щодо чітко визначених структурних або метаболічних розладів, деякі з яких також мають генетичні причини. Вважається, що більше половини випадків епілепсії мають генетичну основу [1]. Застосування геномних технологій має величезний вплив на відкриття генетичної етіології епілепсії та відіграє ключову роль у діагностиці та раціональному лікуванні епілепсії.
Однак епілепсії, пов’язані з генетичними аномаліями, демонструють велику гетерогенність. Мутації в деяких генах можуть вибірково викликати епілепсію або синдроми, основними симптомами яких є судоми [2], тоді як інші гени можуть бути пов’язані з грубими вадами розвитку мозку та епілепсією [3, 4]. Разом із цим судоми також можуть виникати при інших генетичних захворюваннях, що впливають на центральну нервову систему [5, 6]. Тому складно вирішити, який ген або групу генів слід охарактеризувати в конкретній цільовій популяції пацієнтів, перш ніж розробити ефективну стратегію генетичного тестування.
У даній статті ми розберемо кілька клінічних випадків, у яких ключову роль відіграло генетичне секвенування в діагностиці рідкісних неврологічних захворювань, та встановимо подальшу тактику ведення таких пацієнтів.
Випадок 1
Дівчинка М. надійшла до неврологічного відділення 3-ї міської дитячої лікарні м. Одеси у віці 5 місяців зі скаргами на відмову від їжі (у першій половині доби ефективного смоктання немає, ближче до вечірнього часу дитина могла з’їсти до 150 мл грудного молока), а також часті відрижки фонтаном, які відзначалися з народження, втрату маси тіла (МТ), млявість, частий плач та занепокоєння, особливо при дотиках та гучних звуках, відкочування у фізичному розвитку (стала гірше тримати голову).
З анамнезу життя відомо, що дитина від 2-ї вагітності, 2-х пологів у терміні 39 тижнів (перша дитина — дівчинка, 4 роки, здорова), МТ при народженні 2860 г, довжина тіла 49 см, оцінка за шкалою Апгар 8–9 балів.
Анамнез захворювання: мати пов’язує дебют захворювання з початком вакцинації (ротарікс, гексаксим), тому що в першу добу після цього відзначалися 4 епізоди блювання фонтаном із подальшою поступовою відмовою від їжі. Динаміка МТ дитини подана в табл. 1.
При оцінці неврологічного статусу встановлено, що дитина млява. Фіксація погляду нестійка. Під час вигодовування відзначаються періодичні дискоординовані рухи очних яблук (підкочування, девіація вбік). Тургор знижений. Виражена гіперестезія. Фонація не порушена. М’язовий тонус дистонічний із переважанням гіпертонусу в нижніх кінцівках. Сила знижена. Під час тракції за руки групується погано. Сухожильні рефлекси з рук та ніг торпідні, симетричні. Клоноїду немає. Рефлекси періоду новонародженості (повзання, автоматичної ходи, Моро) викликаються, пожвавлені, не згасають. При спробі вертикалізації установка нижніх кінцівок із перехрестом, опора еквінусна.
Дитина була консультована лікарем-гастроентерологом, який установив діагноз: «порушення харчової поведінки, диспепсія». При подальшому неодноразовому дослідженні шлунково-кишкового тракту патології не виявлено.
Разом із цим під час знаходження пацієнтки у стаціонарі була проведена низка досліджень, які не виявили змін, а саме: дослідження на TORCH-інфекції, визначення функції щитоподібної залози (ТТГ, Т3, Т4), дослідження електролітів крові, гіпо- та гіпервітамінози, амінокислотний скринінг. Проте відзначалось незначне підвищення лактату крові — 2,4 ммоль/л.
У віці 5 місяців пацієнтці були проведені дослідження: МРТ головного мозку та ЕЕГ-відеомоніторинг. Результати подані в табл. 2.
За час перебування в стаціонарі дитина отримувала метаболічну терапію, вигодовувалась зондом — 60 %, самостійно — 40 %, збільшення у масі тіла за 10 днів 100 г. Після виписки стан дитини в динаміці не покращувався: втрата навичок (перестала фіксувати погляд, утримувати голову, посміхатися), вигодовувалась через зонд, продовжувала рясно зригувати.
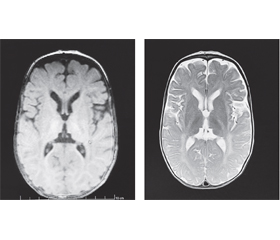
У семимісячному віці дитині було зроблено повторне МРТ-дослідження головного мозку з наступним висновком: на тлі дифузних помірно виражених атрофічних змін паренхіми півкуль головного мозку простежуються великі ділянки гіпо/демієлінізації в ділянці базальних ядер, у перивентрикулярних відділах із поширенням по білій речовині напівовальних центрів до субкортикальних відділів потиличних, тім’яних, дещо менше — скроневих і лобових, «тигроїдного» рисунка. Візуалізується виражене уповільнення дифузії у проєкції кортикоспінальних трактів обох півкуль головного мозку. Крім того, на тлі зменшення в обсязі паренхіми мозочка визначаються симетричні хмароподібні ділянки демієлінізації на рівні середніх ніжок мозочка, біля зубчастих ядер. Потовщення хіазми та проксимальних сегментів зорових нервів. Явища замісної вентрикулодилатації. МР-картина може відповідати нейродегенеративному процесу (МР-картина суспектна синдрому Crabbe та MLD) (рис. 1).
/36.jpg)
Дитині проведене генетичне секвенування (панель лейкодистрофій) та виявлено мутацію в гені GALC у гомозиготній формі, розташованому на 14-й хромосомі, що пов’язана з хворобою Краббе, яка успадковується за автосомно-рецесивним типом. В основі захворювання лежить зниження активності ферменту галактозилцерамід-β-галактозидази, який у нормі розщеплює галактоцереброзид на церамід та галактозу. Відбувається накопичення негідролізованих субстратів у бімолекулярному шарі мієлінового волокна як центральної, так і периферичної нервової системи, викликаючи загибель олігодендроцитів, розпад мієлінового волокна та утворення характерних включень — глобоїдних клітин.
На момент публікації статті дитині М. 1 рік 6 міс., МТ — 5800 г, захворювання неухильно прогресує. Амавроз, псевдобульбарні порушення (вигодовується через зонд). Спастичний тетрапарез. Часто хворіє на вірусно-бактеріальні інфекції, пневмонії.
Хотілось б нагадати колегам, що специфічної терапії при хворобі Краббе немає та прогноз несприятливий. Основним методом лікування є алогенна трансплантація кісткового мозку (або пуповинної крові), яку рекомендовано проводити до розвитку помітних неврологічних порушень на ранніх етапах захворювання, оскільки трансплантація не може вплинути на пошкодження ЦНС, що вже виникли, проте може зупинити або уповільнити їх подальше прогресування.
Внутрішньоутробно хворобу Краббе можна діагностувати за допомогою пренатального скринінгу — біопсії ворсин хоріону або амніоцентезу для визначення активності ферменту галактоцереброзидази.
Випадок 2
Дитина М., дівчинка віком 7 міс., надійшла у неврологічне відділення зі скаргами на частковий двобічний птоз, стридорозне дихання, втрату фізичних навичок.
Анамнез життя: дитина від 3-ї вагітності, 2-х пологів (1-ша вагітність — мимовільний аборт на ранніх термінах; друга вагітність — здорова дівчинка, 4 роки), у терміні 39 тижнів, із МТ 3300 г. Перинатальний анамнез не обтяжений. За словами матері, до 4 місяців розвивалася згідно з віком, стридор — із народження.
Анамнез хвороби: мати відзначає повільний регрес у психоемоційному та фізичному розвитку протягом останніх 2 місяців, після того, як дитина перенесла ГРВІ.
При надходженні до стаціонару неврологічний статус: стан дитини тяжкий за рахунок загальномозкової та вогнищевої симптоматики. Млява, адинамічна, сонлива. Очні щілини рівні, частковий двобічний птоз. Фотореакції живі, симетричні. Погляд фіксує нестійко. М’язовий тонус та сила знижені. Голову не тримає, не перевертається, іграшку до рук не бере. Сухожильні рефлекси живі, симетричні. Менінгеальних знаків немає.
При проведенні МРТ-дослідження головного мозку встановлено: ознак об’ємних змін головного мозку не виявлено. Розширення підпавутинних лікворних просторів. Симетричні зміни МР-сигналу від базальних гангліїв (медіальна бліда куля) та чорної субстанції ніжок мозку.
Під час перебування на стаціонарному лікуванні дитина отримувала дегідратаційну та метаболічну терапію, після чого дівчинка стала активнішою — краще почала тримати голову, з’явився захват іграшки кистю, зменшився птоз. Дитину виписано додому з позитивною динамікою через 2 тижні.
Через 1 місяць мати звернулася повторно у зв’язку з наростаючим порушенням функції зовнішнього дихання (посилення стридору, що супроводжується вираженим втягуванням допоміжної мускулатури, ціанозом носогубного трикутника і падінням сатурації до 88 % під час плачу), гастроінтестинальними порушеннями і затримкою психомоторного розвитку (частковий птоз, груба дифузна м’язова гіпотонія, відсутність зорового контакту).
Повторно проведено МРТ мозку — зберігаються зміни МР-сигналу від базальних гангліїв та середнього мозку, порівняно з попереднім станом — тенденція до регресу.
Проведене генетичне секвенування, виявлена мутація в гені SCO2 у гомозиготній формі. Цей ген пов’язаний з автосомно-рецесивною кардіоміоенцефалопатією, спричиненою дефіцитом мітохондріального комплексу IV. Це автосомно-рецесивне тяжке мітохондріальне захворювання, при якому порушується енергетичний обмін у всіх життєво важливих органах (серце, легені, мозок).
Ефективного методу лікування цього захворювання нині немає. Стан дитини прогресивно погіршувався. На жаль, у віці 9 місяців дитина померла від кардіореспіраторної недостатності.
Випадок 3
Хлопчик віком 1 рік 5 міс. надійшов до неврологічного стаціонару зі скаргами на часті серійні напади судом у вигляді синхронних, симетричних, раптових посмикувань верхнього плечового пояса (руки зігнуті в ліктьових суглобах, при кожному посмикуванні — ступінчасто піднімаються вгору).
Анамнез захворювання: уперше напади з’явилися у віці 1 рік 1 міс. із частотою до 3 серій на день (3–4 напади у серії). Поява нападів часто провокувалася емоційним збудженням дитини (переляк, хвилювання, плач). За 2 тижні до початку — друга ревакцинація АКДС.
Частота нападів поступово зростала, збільшувалася кількість нападів у серії (до 15–20). Ці стани трактувалися як емоційні реакції. Дитину не було обстежено, терапію не отримувала. У 1 рік 3 міс. дитина перенесла ентеровірусну інфекцію (вірус Коксакі), на тлі чого напади стали мати статусний характер. Дитина стала збудженою, дратівливою. Наголошувалося на втраті ефективної комунікації через збіднення емоційного фону.
При надходженні до стаціонару дитину було обстежено. На рис. 2 подано ЕЕГ-дослідження при госпіталізації.
З ЕЕГ-дослідження видно, що генералізовані спалахи комплексів поліпік-хвиля 2,5–3 Гц, амплітуда 240–385 мкВ, з акцентом у лобовій ділянці, тривалістю 7–8 секунд.
Разом із цим було проведено МРТ головного мозку з наступним висновком: МР-ознак об’ємних та патологічних вогнищевих змін головного мозку не виявлено. Ретроцеребелярна арахноїдальна кіста.
Під час перебування дитини в стаціонарі 3-ї міської дитячої лікарні м. Одеси було встановлено діагноз: ідіопатична генералізована епілепсія з міоклонічними абсансами. Згідно з протоколом лікування при міоклонічних нападах призначена перша лінія протисудомних препаратів: леветирацетам, вальпроат натрію, топірамат.
Першочергово був уведений препарат вальпроєвої кислоти (з поступовим збільшенням дози до 20 мг/кг/добу). На тлі прийому вальпроату частота нападів не змінилася, відзначалась виражена побічна дія на психоемоційну сферу дитини (млявість, апатія). Поступово введено леветирацетам (30 мг/кг/добу). Кількість нападів зменшилась, серії стали коротшими.
Через 3–4 дні прийому леветирацетаму частота нападів знову зросла (постійні протягом доби, у серії до 20 нападів). Збільшено дозування до 40 мг/кг/добу. Частота нападів суттєво не змінилася. Був доданий клоназепам (0,1 мг/кг/добу). Відзначалася позитивна динаміка: протягом 5 днів напади залишалися тільки при засинанні та при психоемоційному збудженні (до 5–7 у серії).
З указаними вище рекомендаціями, а саме прийом вальпроату, леветирацетаму, клоназепаму, дитина була виписана додому.
Через 10 днів після виписки дитина захворіла на коронавірусну інфекцію (COVID-19), частота нападів знову зросла до 200 на добу. Напади постійні, до 20–25 у серії. Дитина млява, складно доступна контакту. Госпіталізована до стаціонару.
Хлопчику було проведене генетичне обстеження: виявлено мутацію гена CACNA1H c.1912G>A (p.Gly638Ser), гетерозиготну форму.
Нагадаємо, що ген CACNA1H (англ. Calcium voltage-gated channel subunit alpha1 H) розташований на хромосомі 16 та кодує білок, який функціонально відноситься до низькопорогових кальцієвих каналів. Він утворює пору в клітинній мембрані, за допомогою якої транспортує іони кальцію в клітину електрохімічним градієнтом. У нейронах кальцієві канали регулюють збудливість клітинної мембрани. Ця мутація асоційована з автосомно-домінантною генералізованою епілепсією.
Ґрунтуючись на результатах генетичного дослідження, була проведена заміна вальпроату на топірамат, враховуючи його дію на кальцієві канали (рис. 3).
На момент публікації статті дитині 1 рік 8 міс., зберігаються поодинокі напади у вигляді коротких серій до 3–5 легких посмикувань при засинанні 1–2 рази на тиждень. Дитина активна, емоційна, контактна. Прий-має леветирацетам 40 мг/кг/добу, топірамат 5 мг/кг/добу. На ЕЕГ також відзначається позитивна динаміка: реєструється поліморфна активність, схильність до депресії ритму, амплітуда 8–25 мкВ, зональні відмінності відсутні (рис. 4).
Висновки
За результатами наших спостережень з яскравими клінічними проявами ми можемо зробити такі висновки:
1) метод генетичного обстеження є одним із провідних у диференціальній діагностиці епілепсії;
2) за допомогою генетичного секвенування ми маємо змогу встановити етіологію захворювання;
3) разом із вищевказаними перевагами ми можемо надавати ефективну таргетну допомогу нашим маленьким пацієнтам;
4) ці дані є новим підґрунтям для подальших досліджень.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 14.01.2022
Рецензовано/Revised 31.01.2022
Прийнято до друку/Accepted 03.02.2022

/35.jpg)
/36.jpg)
/37.jpg)
/38.jpg)
/38_2.jpg)