Вступ
Кінцівково-поясна м’язова дистрофія (LGMD) — це неоднорідна група генетично обумовлених нервово-м’язових захворювань, що характеризуються прогресуючою слабкістю і атрофіями м’язів тазового та плечового поясів, а також значно варіабельним перебігом захворювання [1]. Найчастіше при даних формах м’язових дистрофій уражуються м’язи плечей, верхніх кінцівок, тазового пояса та стегон [2, 3].
Найбільш ранні описи пацієнтів з прогресуючою слабкістю м’язів кінцівково-поясної групи належать відомим німецьким лікарям Ернсту Віктору фон Лейдену (1876) та Паулю Юліусу Мебіусу (1879). Вони вперше описали дорослих пацієнтів з слабкістю і атрофіями м’язів стегон та тазового пояса.
У 1884 р. німецький невролог Вільгельм Хайн-ріх Ерб у роботі «Ueber die «Juvenile Form» der progressiven Muskelatrophie und ihre Beziehungen zur sogenannten Pseudohypertrophie der Muskeln» вперше описав ювенільну форму проксимальної м’язової слабкості. У 1891 р. В. Ерб сформував концепцію м’язових дистрофій як розладів, що викликаються первинною дегенерацією м’язів, та запропонував термін «dystrophia muscularis progressiva». У східноєвропейській науковій школі дослідження кінцівково-поясної м’язової дистрофії пов’язано з ім’ям видатного російського невролога Володимира Карловича Рота (1848–1916), відомого, серед іншого, такими роботами, як «Патогенез мышечных атрофий» и «О патогенезе прогрессивной мышечной атрофии» (в «Трудах IV съезда Общества русских врачей», 1891). Таким чином, у західній науковій літературі кінцівково-м’язова дистрофія відома під назвою «м’язова дистрофія Ерба», а у східній — «м’язова дистрофія Ерба — Рота».
На сьогодні описано 24 автосомно-рецесивні форми (LGMD2) та 8 автосомно-домінантних (LGMD1) форм [3]. Найбільше страждають м’язи проксимальних відділів кінцівок, особливо м’язи плечей, передпліччя та тазової ділянки [4].
Симптоми зазвичай починаються протягом перших 20 років життя, мають прогресуючий перебіг, що призводить до втрати здатності ходити між 10 і 20 роками після появи перших проявів [5].
М’язова слабкість і втомлюваність у м’язах плечового пояса можуть спричиняти зміни постави та зовнішнього вигляду плечей, спини та рук. Слабкість м’язів плечового пояса призводять до розвитку симптому, відомого як «крилоподібні лопатки» [3–5].
У пацієнтів з LGMD також можуть спостерігатися деформації хребта, включаючи лордоз та сколіоз. У деяких пацієнтів виникають контрактури, які можуть обмежувати рухи у стегнових, колінних, гомілкових та ліктьових суглобах. Псевдогіпертрофії литкових м’язів також можуть зустрічатися у деяких пацієнтів з LGMD [6, 7].
На початку захворювання пацієнти можуть мати незвичну ходу, таку як розкачування або ходьба на носках, можуть виникнути труднощі з бігом. Типовим симптомом у дітей є застосування прийому Говерса при вставанні з підлоги [3, 8].
Діагностичні критерії кінцівково-м’язових дистрофій включають підвищену активність креатинфосфокінази в сироватці крові, зміни на електроміограмі, що вказують на наявність міопатії, дані біопсії м’язів, що виявляють міопатичні або дистрофічні зміни та відсутність або зменшення продукції білка, специфічного для конкретної форми LGMD, за даними вестерн-блоттингу [4].
У деяких пацієнтів з LGMD може розвиватися кардоміопатія. Також можуть виникати різної тяжкості проблеми з диханням, від легких до тяжких, що пов’язано з прогресуючою слабкістю дихальних м’язів. Інтелект у пацієнтів з LGMD, як правило, не страждає, однак затримка розвитку і розумова відсталість були зареєстровані у рідкісних випадках [10, 12, 13].
Тривалість життя у пацієнтів зазвичай знаходиться в межах норми, оскільки серце та дихальні м’язи лишаються неураженими. На пізніх стадіях можуть виникати розлади дихання, але у типових випадках менш тяжкі, ніж при інших м’язових дистрофіях. До симптомів порушення дихання під час сну можна віднести неспокійний поверхневий сон, кошмари, втому або головний біль після пробудження вранці, відсутність апетиту та сонливість протягом дня [3, 10–12].
Поширеність LGMD оцінюється від 1 на 14 500 до 1 на 123 000 осіб [7]. LGMD2A є однією з найбільш частих форм. У європейських країнах поширеність оцінюється як 1 випадок на 100 000 осіб. Поширеність LGMD та окремих форм даної патології в Україні невідомі, тому що не було проведено епідеміологічних досліджень [10].
На сьогодні не існує специфічних методів лікування LGMD2A, однак ретельне спостереження пацієнта та менеджмент симптомів цього захворювання може значно покращити якість життя. Ведення активного способу життя важливо для всіх пацієнтів, які страждають на цю м’язову дистрофію. Немає чітких рекомендацій щодо виду чи інтенсивності фізичної активності, дозволеної пацієнтам, проте рекомендується будь-які вправи виконувати в межах можливостей і комфорту пацієнта. Сильна втома, біль та судоми у м’язах під час або після занять можуть вказувати на перевтому, у цьому випадку необхідно зменшити інтенсивність навантажень. Плавання є оптимальним видом фізичних навантажень, оскільки сприяє активності всіх груп м’язів без перенапруження [14–16].
Як ілюстрацію ми наводимо два клінічні випадки пацієнток з підтвердженим діагнозом кінцівково-поясної м’язової дистрофії типу 2А.
Випадок 1
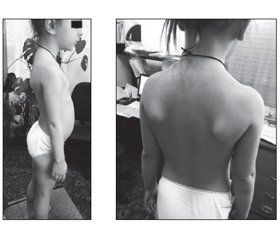
Дівчинка М., 5 років, уперше була обстежена у зв’язку з поступово прогресуючою м’язовою слабкістю у нижніх кінцівках, труднощами з підніманням сходами, вставанням з підлоги та порушеннями ходи. Ці симптоми, зі слів батьків, спостерігалися з 4-річного віку. Також поступово виникли труднощі з утриманням важких предметів в руках та рук над головою (рис. 1–3).
/30.jpg)
Дівчинка народилася від неспорідненого шлюбу. У родині спадкові захворювання не відмічалися. Народжена від 2-ї неускладненої вагітності та термінових фізіологічних неускладнених пологів. Маса тіла при народженні становила 4600 г, оцінка за шкалою Апгар — 8/9 балів. Дівчинка є другою дитиною у родині та має здорового старшого брата і двох молодших братів. Рухові навички в ранньому дитинстві формувалися нормально, сиділа з 7,5 міс., самостійна хода з 12 міс. До 4 років на диспансерному обліку у невролога не перебувала. У віці 5 років під час обстеження після перенесеної респіраторної інфекції випадково виявлено підвищення рівня трансаміназ. Після цього дитина обстежена гепатологом, виключено автоімунні та вірусні гепатити. З метою уточнення діагнозу дитину було направлено до неврологічного відділення ДУ «ІПАГ ім. акад. О.М. Лук’янової НАМНУ».
/30_2.jpg)
Неврологічне обстеження показало виражену симетричну слабкість кінцівок (оцінка м’язової сили 0/5 у дельтоподібних м’язах, 1/5 у біцепсах та трицепсах, 3/5 у розгиначах зап’ястя, 4/5 у згиначах зап’ястя, 0/5 у псоасах, 1/5 у привідних та відвідних м’язах стегон, 2/5 у квадрицепсах, задніх стегнових та великогомілкових м’язах та 4/5 у згиначах стоп), а також виражену м’язову атрофію у проксимальних відділах верхніх та нижніх кінцівок. М’язовий тонус кінцівок був знижений переважно в проксимальних відділах нижніх кінцівок та менш виражено у проксимальних відділах верхніх кінцівок. Також у дитини відмічалися псевдогіпертрофії литок, крилоподібні лопатки та симптом Говерса. Очні, лицеві, бульбарні та шийні м’язи були інтактними. Усі глибокі сухожильні рефлекси були відсутні. На момент огляду не виявлено фасцикуляцій, ознак ураження верхніх рухових нейронів або контрактур суглобів. Чутливість (поверхнева, больова, вібраційна та пропріо-чутливість) також була не порушеною. Інтелектуальний та мовленнєвий розвиток дитини відповідав віку. Рівень КФК у сироватці крові був підвищеним (5500–6000 Од/л). Відмічалося підвищення рівня АЛТ та АСТ до 2 норм. Інші показники біохімії крові, загальних аналізів крові та сечі були в нормі. Форсована життєва ємність легень становила 72 % у сидячому положенні. Даних щодо патології під час ЕКГ, ехокардіографії (ЕхоКГ) та рентгенографії грудної клітки не виявлено. Соматичний статус дитини був без істотних відхилень.
Проведено обстеження батьків та рідних братів пацієнтки, у всіх родичів виявлено нормальний рівень КФК та відсутність симптомів м’язової слабкості у кінцівках або порушень ходи.
Беручи до уваги наявність у дитини прогресуючої м’язової слабкості та атрофій, переважно у проксимальних відділах верхніх та нижніх кінцівок, підвищення рівня КФК, була запідозрена наявність прогресуючої м’язової дистрофії. Для уточнення форми захворювання батькам дитини було запропоновано проведення генетичного дослідження.
Аналіз послідовностей та тестування на делеції/дуплікації 159 генів, пов’язаних із захворюваннями скелетних м’язів, виявив делецію екзону 8 (у гетерозиготному стані) та мутацію c5050delA (p.Thr184Argfs*36) (у гетерозиготному стані) у гені CAPN3. Дані мутації гена CAPN3 є патогенними та асоціюються з автосомно-рецесивною кінцівково-поясною м’язовою дистрофією 2А (LGMD2A) (MedGen UID: 358391).
Було проведено МРТ м’яких тканин проксимальних відділів нижніх кінцівок, яке показало ознаки симетричних атрофічних змін великого привідного м’яза, довгого і короткого привідного м’язів, півсухожильного м’яза стегна як проявів прогресуючої м’язової дистрофії (рис. 4).
Випадок 2
Дівчинка Г., 9 років, уперше госпіталізована до педіатричного відділення ДУ «ІПАГ ім. акад. О.М. Лук’я-нової НАМНУ» у 8 років зі скаргами на втомлюваність, зниження апетиту, відчуття дискомфорту та слабкості у нижніх кінцівках.
З анамнезу відомо, що дитина росла та розвивалася згідно з віком, вакцинована згідно з календарем, операцій, травм, тяжких інфекційних захворювань в анамнезі немає. Скарги з’явилися за 2 місяця до госпіталізації, коли у дівчинки відмічалося кількаразове блювання. Була госпіталізована до стаціонару приватної клініки, де виявлено підвищення рівня транс-аміназ. Консультована інфекціоністом, виключено вірусні гепатити В та С, виявлені IgG до гепатиту А. Після виписки дитина отримала курс протигельмінтної терапії, після якої повторно виявлено підвищення рівня трансаміназ та підвищення рівня креатинфосфокінази у сироватці крові (5801 Од/л). Дитина направлена на дообстеження до педіатричного відділення ДУ «ІПАГ ім. О.М. Лук’янової НАМНУ». На момент госпіталізації стан дитини був середньої тяжкості у зв’язку із симптомами м’язової слабкості та втомлюваності.
Під час огляду дівчинка активна, суб’єктивно відмічає слабкість у м’язах нижніх кінцівок, переважно у стегнах. Скаржиться на відчуття тяжкості і втоми у нижніх кінцівках при ходінні. Контакту доступна, на огляд реагує спокійно. Когнітивний та мовленнєвий розвиток дитини відповідає віку. Астенічної тілобудови. Наявна сколіотична постава, крилоподібні лопатки (рис. 5). Зіниці округлої форми, симетричні. Фотореакція жива, співдружня. Ністагм відсутній. Мімічна іннервація симетрична. Ковтання, фонація не порушені. Черевні рефлекси живі, симетричні. М’язовий тонус знижений у плечовому поясі та нижніх кінцівках. Помірно виражена симетрична слабкість кінцівок (оцінка м’язової сили 2/5 у дельтоподібних м’язах, 2/5 у біцепсах та трицепсах, 3/5 у розгиначах зап’ястя, 4/5 у згиначах зап’ястя, 3/5 у псоасах, 2/5 у привідних та відвідних м’язах стегон, 3/5 у квадрицепсах, задніх стегнових та великогомілкових м’язах та 4/5 у згиначах стоп), а також виражена м’язова атрофія у проксимальних відділах верхніх та нижніх кінцівок. Хода не порушена. Сухожильні рефлекси знижені, переважно у нижніх кінцівках, D = S. Координаторні проби виконує правильно. Патологічні рефлекси відсутні. Розлади чутливості відсутні. Порушень функцій тазових органів немає. Відмічаються труднощі при вставанні після присідання навпочіпки, симптом Говерса. Відмічаються труднощі з підніманням сходами.
/31.jpg)
У відділенні проведено комплексне обстеження. Загальноклінічні аналізи крові та сечі без відхилень. Біохімічне дослідження крові виявило підвищення рівня трансаміназ (АЛТ — 274 Од/л, АСТ — 289 ОД/л), загальної креатинфосфокінази (4662 Од/л), зниження рівня вітаміну D3 (25-гідроксикальциферол — 54,3 ммоль/л).
Ультразвукове дослідження органів черевної порожнини: ознаки дифузних змін паренхіми печінки.
Еластографія печінки: жорсткість печінки F0 за METAVIR.
Ехокардіографія: порожнини серця не збільшені. Структура і функція клапанів не порушені. Скоротливість міокарда добра. Вільної рідини в обох плевральних порожнинах немає.
МРТ серця з контрастуванням: ознак структурних змін серця не виявлено. Вільної рідини в порожнині перикарда не виявлено.
Комп’ютерна томографія органів черевної порожнини із заочеревинним простором та малого таза: ознаки помірно вираженої гепатомегалії. Даних щодо вогнищевої та об’ємної патології під час дослідження не виявлено.
Електроміографія м’язів: ознак первинно-м’язового, мотонейронального ураження не виявлено.
Дитина консультована ревматологом: переконливих даних щодо наявності ревматичних захворювань під час обстеження не виявлено.
Зважаючи на клінічні прояви міопатичного синдрому, підвищення рівня трансаміназ та креатинфосфокінази, дитина була консультована неврологом. Попередній діагноз: прогресуюча м’язова дистрофія.
За результатами діагностичного тестування у лабораторії «INVITAE» виконали аналіз секвенування та тестування на делеції/дуплікації 153 генів (панель кардіоміопатії та скелетно-м’язових захворювань). Результат дослідження позитивний, виявлено два патогенних варіанти в CAPN3, CAPN3 асоційований з автосомно-рецесивною та домінантною кінцівково-поясною м’язовою дистрофією.
З урахуванням даних анамнезу, клінічного обстеження та результатів генетичного тестування було встановлено діагноз кінцівково-поясної м’язової дистрофії, тип 2А, для підтвердження якої пацієнтку було направлено до медико-генетичного центру НДСЛ «ОХМАТДИТ» МОЗ України.
Дитина консультована в медико-генетичному центрі, де підтверджено попередній діагноз, встановлений неврологом: кінцівково-поясна м’язова дистрофія, тип 2А. Дифузне захворювання печінки.
Висновки
Прогресуючі м’язові дистрофії — одна з важливих проблем в дитячій неврології. Важливо своєчасно розпізнавати прояви м’язової дистрофії у дітей, особливо враховуючи те, що діти з даною патологією нерідко потрапляють до педіатрів у зв’язку з підвищенням рівня печінкових ферментів. Характерною ознакою для всіх м’язових дистрофій є двостороння симетричність ураження скелетної мускулатури, м’язова атрофія та слабкість, що ведуть до порушення моторних і статичних функцій. Особливе місце серед прогресуючих м’язових дистрофій посідає група кінцівково-поясних м’язових дистрофій.
Пацієнти з дистрофією Ерба — Рота потребують комплексного клініко-інструментального підходу із залученням суміжних спеціалістів. Зважаючи на неврологічну симптоматику, лабораторні та інструментальні дослідження, можна виділити діагностичні критерії даного захворювання, що включають підвищену активність креатинфосфокінази та трансаміназ у сироватці крові, зміни на електроміограмі, що вказують на наявність міопатії, дані біопсії м’язів, що виявляють міопатичні або дистрофічні зміни, та відсутність або зменшення продукції білка, специфічного для конкретної форми кінцівково-поясної м’язової дистрофії за даними вестерн-блоттингу, а також атрофічні зміни за даними МРТ м’язів. На підставі генетичного дослідження можна підтвердити діагноз шляхом виявлення мутацій генів, характерних для кінцівково-поясних м’язових дистрофій, методом секвенування наступної генерації (дослідження панелі генів або повноекзомне секвенування). На сьогодні не існує спеціальних методів лікування кінцівково-поясної м’язової дистрофії, проте ретельний менеджмент симптомів може суттєво поліпшити якість життя дитини та уповільнити прогресування захворювання.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 18.01.2022
Рецензовано/Revised 31.01.2022
Прийнято до друку/Accepted 10.02.2022
Список литературы
1. Cotta A., Carvalho E., da-Cunha-Júnior A., Paim J., Navarro M., Valicek J. et al. Common recessive limb girdle muscular dystrophies differential diagnosis: why and how? Arquivos de Neuro-Psiquiatria. 2014. 72(9). 721-734. doi: https://doi.org/10.1590/0004-282X20140110.
2. Albuquerque M. Limb-girdle muscular dystrophy in Brazilian children: clinical, histological and molecular characterization. Arquivos de Neuro-Psiquiatria. 2014. 72(6). 481-481. doi: https://doi.org/10.1590/0004-282x20140037.
3. Angelini C., Nardetto L., Borsato C., Padoan R., Fanin M., Nascimbeni A. et al. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurological Research. 2010. 32(1). 41-46. doi: https://doi.org/10.1179/174313209X380847.
4. Broglio L., Tentorio M., Cotelli M., Mancuso M., Vielmi V., Gregorelli V. et al. Limb-Girdle Muscular Dystrophy-Associated Protein Diseases. The Neurologist. 2010. 16(6). 340-352. doi: https://doi.org/10.1097/NRL.0b013e3181d35b39.
5. Dincer P. A cross section of autosomal recessive limb-girdle muscular dystrophies in 38 families. Journal of Medical Genetics. 2000. 37(5). 361-367. doi: https://doi.org/10.1136/jmg.37.5.361.
6. Grimm D., Hanisch F., Liu X., Müller-Reible C., Zierz S., Deschauer M. Frequency of calpain-3 c.550delA mutation in limb girdle muscular dystrophy type 2 and isolated hyperCKemia in German patients. Aktuelle Neurologie. 2006. 33(S 1). doi: https://doi.org/10.1055/s-2006-95302211.
7. Haberlová J. Evaluation and Treatment of Myopathies. European Journal of Human Genetics. 2015. 23(10). 1433-1433. doi: https://doi.org/10.1038/ejhg.2015.127.
8. Kaplan J., Hamroun D. The 2015 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscular Disorders. 2014. 24(12). 1123-1153. doi: https://doi.org/10.1016/j.nmd.2014.11.001.
9. Khadilkar S. Limb girdle muscular dystrophies in India. Neurology India. 2015. 63(4). 495. doi: https://doi.org/10.4103/0028-3886.161989.
10. Liu W., Pajusalu S., Lake N., Zhou G., Ioannidis N., Mittal P. et al. Estimating prevalence for limb-girdle muscular dystrophy basedon public sequencing databases. Genetics in Medicine. 2019. 21(11). 2512-2520. doi: https://doi.org/10.1038/s41436-019-0544-8.
11. Oliveira Santos M., Ninitas P., Conceição I. Severe limb-girdle muscular dystrophy 2A in two young siblings from Guinea-Bissau associated with a novel null homozygous mutation in CAPN3 gene. Neuromuscular Disorders. 2018. 28(12). 1003-1005. https://doi.org/10.1016/j.nmd.2018.09.009.
12. Pegoraro E., Hoffman E.P. Limb-Girdle Muscular Dystrophy Overview. 2000 Jun 8 [Updated 2012 Aug 30]. In: Adam M.P., Ardin-ger H.H., Pagon R.A. et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 1993–2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1408/
13. Stöllberger C., Finsterer J. Cardiopulmonary involvement in limb girdle muscular dystrophy 2A. Muscle & Nerve. 2017. 56(4). E38-E38. doi: https://doi.org/10.1002/mus.25369.
14. Wang C., Liang W., Minami N., Nishino I., Jong Y. Limb-girdle Muscular Dystrophy Type 2A with Mutation in CAPN3: The First Report in Taiwan. Pediatrics & Neonatology. 2015. 56(1). 62-65. doi: https://doi.org/10.1016/j.pedneo.2013.01.018.
15. Genetics Home Reference. Limb-girdle muscular dystrophy. US National Library of Medicine. Available at: https://ghr.nlm.nih.gov/condition/limb-girdle-muscular-dystrophy. 2014 Dec; Accessed: Aug 8, 2019.
16. Piluso G., Politano L., Aurino S., Fanin M., Ricci E., Ventriglia V.M. et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J. Med. Genet. 2005. 42. 686-693.

/30.jpg)
/30_2.jpg)
/31.jpg)