Останніми десятиліттями актуальність проблеми прогнозування перинатального гіпоксично-ішемічного ушкодження (ГІУ) центральної нервової системи (ЦНС) у новонароджених різного гестаційного віку стала більш очевидною у зв’язку із збільшенням кількості дітей, які народилися раніше терміну, та збільшенням їх виживання. Проте перинатальна патологія ЦНС є не тільки медичною, а й соціальною проблемою через високу інвалідизацію цієї групи дітей [1, 4, 7, 9, 50].
Згідно зі статистичними даними Міністерства охорони здоров’я України, показник дитячої інвалідності в Україні за останні 5 років зріс на 7,8 % і на 01.01.2020 року становив 163 886 дітей. Серед причин інвалідності в дітей віком до 18 років на першому місці виокремлено вади розвитку, деформації та хромосомні аномалії — 49 206 дітей (питома вага 30,0 %), на другому місці — розлади психіки та поведінки — 27 052 дитини (питома вага 16,5 %), на третьому — захворювання нервової системи — 25 422 дитини (питома вага 15,3 %). Отже, у понад 100 тисяч дітей основною причиною інвалідності, як безпосередньою, так і дотичною, є патологія нервової системи.
Проте частоту перинатальних ушкоджень мозку не можна вважати однозначно встановленою. Це пов’язано з нечіткістю діагностичних критеріїв неврологічної норми й патології, наявністю перехідних (транзиторних) станів, пов’язаних з явищами постнатальної адаптації в новонароджених у перший місяць життя, особливо в передчасно народжених дітей [3, 5, 8, 63].
Сучасні методики нейровізуалізації, які грунтуються на різних біофізичних принципах отримання зображення мозкових структур, такі як нейросонографія, комп’ютерна томографія, магнітно-резонансна томографія, дають змогу виявляти різні зміни з боку структур головного мозку, починаючи з перших днів життя. Однак подальші дослідження і спостереження за дітьми з перинатальною патологією ЦНС вказують на те, що обсяг і характер змін, виявлених даними методиками в неонатальному періоді, не завжди корелюють із вираженістю неврологічних відхилень, які в подальшому формуються, що не дає змогу використовувати їх як надійні прогностичні критерії [14, 31].
Типові патобіохімічні й патофізіологічні процеси, що формують основу перинатального ГІУ ЦНС, складаються з порушення нейротрансмітерної й нейротрофічної сигналізації, електрозбудженості, порушення нейронгліальних взаємодій й енергетичного метаболізму, нейрозапалення, некрозу, апоптозу і дизрегуляції нейрогенезу. Водночас зміни, які виникають у клітинах нервової тканини в постнатальному періоді, залишаються недостатньо вивченими. Нині ведеться пошук ранніх маркерів ушкодження клітин головного мозку, досліджуються можливі шляхи захисту від агентів, що ушкоджують, а також способи активації репаративних процесів [2, 32].
До потенційних напрямків ранньої діагностики ушкодження ЦНС належить виявлення нейромаркерів — специфічних продуктів деградації тканини ней-рональної й гліальної природи, а також метаболітів і нейротрансмітерів, рівень яких змінюється в крові й лікворі під впливом гіпоксії, ішемії та запалення [20, 32, 59].
Нейроспецифічні білки — це група білків, які є специфічними для клітин нервової тканини. Вони відіграють важливу роль у координації внутрішньоклітинних процесів і міжклітинних взаємодій, регуляції функціонального дозрівання і морфологічного розвитку ЦНС. Реєструючи вміст нейроспецифічних білків у біологічних зразках (кров, ліквор), можна підтвердити факт ушкодження ЦНС і визначити його вираженість. Ідентифіковано кілька десятків таких білків: гліофібрилярний кислий білок (Glial fibrillary acidic protein — GFAP) і білок S-100β, що експресується астроглією; основний білок мієліну (myelin basiс protein — MBP, ОБМ) — один із білків мієлінової оболонки; враховуючи те, що у формуванні та підтримці миєлінового шару бере участь олігодедроглія, його відносять до гліальних маркерів); нейронспецифічна єнолаза (NSЕ) та убіквітин-С-кінцева гідролаза L1 (UCHL1) — маркери ушкодження нейронів; sPECAM1, sICAM1 — маркери ендотеліальної дисфункції та ін. [20, 21, 32, 59, 64L].
Асептичне запалення як один із механізмів, що підсилює і пролонгує процес ушкодження мозку, багато в чому пов’язане з багатофункціональним прозапальним цитокіном, що утворюється переважно моноцитами і макрофагами, — фактором некрозу пухлини альфа (TNF-α).
Важливим є вивчення компенсаторних механізмів, що впливають на перебіг та наслідки гіпоксично-ішемічного ушкодження мозку в дітей, а також визначення стану системи трофічного захисту мозку в неонатальному періоді. В експериментальних роботах (Bennett D.L., Hill C.A., Fitch R.H., Голосної Г.І. та ін.) встановлено, що саме баланс у системі трофічних і ростових факторів забезпечує можливість збереження тканин мозку в критичні моменти [2, 16, 35].
В основі ГІУ ЦНС у новонародженої дитини, зокрема передчасно народженої, лежать порушення метаболічних процесів у клітинах головного мозку (метаболічна енцефалопатія) [6, 15, 18, 27].
Пусковим механізмом «метаболічної катастрофи» є дефіцит кисню й енергетичний стрес, а безпосередніми факторами, що ушкоджують мозок, є продукти порушеного метаболізму. У мозку новонароджених, які перенесли гіпоксію в перинатальному періоді, через метаболічні розлади відбувається накопичення, а рідше дефіцит тих чи інших біохімічних субстратів, які можна кваліфікувати як маркери ушкодження мозку.
Отже, схема нейрональних втрат може бути така: зниження мозкового кровотоку і PO2 → порушення метаболізму глюкози (гексозомонофосфатний шлях) → порушення синтезу ліпідів і нуклеїнових кислот → зниження pH тканин (у периваскулярному просторі) → накопичення молочної кислоти і підвищення pCO2 → порушення гомеостазу кальцію і зниження високоенергетичних фосфатних сполук → підвищення рівня лактату в тканинах мозку → накопичення жирних кислот (арахідонової кислоти) → зміна проникності мембрани нейронів → активація перекисного окиснення ліпідів → гіперпродукція оксиду азоту (NO) → мітохондріальна дисфункція → загибель клітини (некроз/апоптоз) [10, 12].
Базовими патофізіологічними механізмами ГІУ ЦНС є системна гіпоксемія (зниження парціального тиску кисню в артеріальній крові [PO2]) та ішемія (зниження рівня мозкового кровотоку) в мозку [29, 33, 57, 58]. Із цього боку механізми ушкодження мозку в новонароджених схожі на механізми ушкодження в дорослих, хоча в новонароджених переважають інші етіологічні чинники, які призводять до запуску ушкодження структур головного мозку.
Як відомо, у патогенезі ГІУ ЦНС виділяють декілька фаз ушкодження мозку [11, 22, 25, 34].
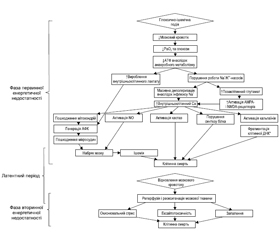
Фаза первинного ушкодження розвивається в момент критичного зниження мозкового кровотоку і парціального тиску кисню (PO2), що супроводжується загибеллю клітин головного мозку упродовж перших годин після цього впливу й обумовлено первинною енергетичною недостатністю. Первинна недостатність енергії спочатку призводить до зменшення рівня кисню і глюкози, а потім до зниження рівня аденозинтрифосфату (АТФ) і підвищення рівня лактату. Низький рівень АТФ спричиняє припинення функціонування багатьох механізмів, що підтримують цілісність клітин, зокрема натрій-калієвих (Na/K) насосів. Неефективність роботи Na/K-насосів призводить до надмірного надходження натрію і кальцію всередину клітини, масивної деполяризації мембрани нейронів, що, зі свого боку, забезпечує вивільнення глутамату. Глутамат, пов’язуючись із NMDA- (N-метил-D-аспартат) і AMPA- (α-аміно-3-гідрокси-5-метил-4-ізоксазолпропіонова кислота) рецепторами, активує їх, посилюючи таким чином додатковий приплив кальцію і натрію в клітину. Порушення натрієвого градієнта призводить до набряку з подальшим лізисом клітини (первинне ушкодження). Надмірна концентрація внутрішньоклітинного кальцію призводить до посилення токсичності накопиченого NO, пошкодження мітохондрій і мікросудин із подальшим наростанням набряку мозку, ішемії та загибелі клітин шляхом некрозу й патологічного апоптозу (рис. 1).
/13.jpg)
Більшість випадків первинної енергетичної недостатності закінчується некрозом клітин. При цьому необернений процес загибелі клітин відбувається в умовах тяжкої гіпоксії й ішемії. При менш тяжкому й розтягнутому в часі ГІУ мозку може бути як відновлення нейронів та їх функцій, так й активація апоптозу — запрограмованої клітинної загибелі. Як відомо, апоптоз викликає ушкодження клітин, не порушуючи при цьому цілісності клітинних мембран. Перевагу одного з цих механізмів чи їх поєднання зумовлюють або тяжкість ушкодження мозку, або відновлювальний потенціал. Первинна енергетична недостатність не завжди призводить до необернених змін [11, 22, 25, 34]. Важливо наголосити на тому, що в переважної більшості випадків вслід за первинною настає вторинна енергетична недостатність, що призводить до подальшої дезінтеграції та дезорганізації роботи мозку.
Між первинною та вторинною фазою енергетичної недостатності є короткий, так званий латентний період — відносного відновлення кровотоку та церебрального метаболізму. Передбачається, що латентний період тим коротший, чим більш тяжкий ступінь ГІУ ЦНС. На сьогодні немає єдиної думки про те, коли закінчуються фаза первинної енергетичної недостатності, коли настає латентний період та коли починається вторинна фаза. На думку деяких дослідників [34], початок латентної фази обчислюється 30–60 хвилинами після ушкодження. Важливо підкреслити, що латентний період вважається найбільш оптимальним для терапевтичних втручань [37].
Більше відомо про фазу вторинної енергетичної недостатності, що реалізується через 6–48 годин [13] після початку розвитку патологічного процесу. Вторинна фаза енергетичної недостатності супроводжується необоротним ушкоджувальним ефектом гіперпродукції збуджуючих нейротрансмітерів і вільних радикалів, асептичним запаленням, а також кінцевим виснаженням запасів фосфатів.
В останніх наукових роботах [25, 30, 34] означена і третя фаза ГІУ ЦНС (Tertiary brain injury (TBI) — третинне ушкодження головного мозку). Дослідники наголошують на тому, що активні патологічні процеси відбуваються протягом тижнів, місяців і навіть років після гіпоксично-ішемічного ушкодження. Вважається, що ця третя фаза містить механізми стійкого запалення й епігенетичних змін, які призводять до подальших несприятливих наслідків, а саме порушень нейрогенезу і синаптогенезу (рис. 2В).
Патогенез перинатального ГІУ ЦНС має свої особливості і складається з декількох фаз [26, 67].
Гіпоксія та зміна мозкового кровотоку. Первинна фаза (рис. 2А) призводить до змін у судинній сітці при порушенні плацентарного кровотоку. Первісною компенсаторною реакцією після перенесеної гіпоксії-ішемії є збільшення мозкового кровотоку й системного транспорту кисню у відповідь на гіпоксію (зниження парціальної напруги кисню [PO2]) і гіперкапнію (збільшення парціальної напруги вуглекислого газу [PСO2]). Це супроводжується перерозподілом серцевого викиду до життєво важливих органів — головного мозку, серця і залоз внутрішньої секреції, зокрема до наднирників. На ранній стадії артеріальний тиск (АТ) підвищується за рахунок збільшення викиду адреналіну. Незважаючи на коливання величин системного АТ, спрацьовує захисний механізм, спрямований на підтримку мозкового кровотоку, що допомагає зберегти церебральну перфузію на постійному рівні (збереження механізмів авторегуляції мозкового кровотоку). У подальшому зрив авторегуляторних механізмів мозкового кровотоку обумовлює наростання патофізіологічних змін у паренхімі головного мозку. У новонароджених, які перенесли мозкову катастрофу, мозковий кровотік стає пасивно залежним від АТ (феномен pressure-passive cerebral blood flow). За умови тривалої ішемії настає виснаження компенсаторних механізмів. Артеріальну гіпертензію змінює гіпотензія, що призводить до зменшення церебральної перфузії, зниження рівня мозкового кровотоку до критичного рівня та зростання внутрішньоклітинного енергодефіциту [23, 67].
Отже, у відповідь на перенесену гіпоксію-ішемію в новонародженого відбувається перерозподіл кровотоку до життєво важливих органів, передусім до головного мозку. За умови тривалої гіпоксії і виснаження компенсаторних механізмів мозковий кровотік зменшується, що і призводить до вторинного ішемічного ушкодження структур головного мозку [26].
/15.jpg)
Гіпоксія й енергодефіцит. Однією зі складових первинної фази є первинний енергодефіцит (рис. 2А). Зі зниженням церебральної оксигенації відбувається зниження рівня кисню в мозку і клітинний енергетичний метаболізм стає залежним від анаеробного метаболізму (анаеробний гліколіз). Нейрональна тканина намагається компенсувати енергодефіцит шляхом активації анаеробного гліколізу, але з огляду на його меншу в 19 разів енергоефективність енергодефіцит продовжує прогресувати. Це призводить до накопичення молочної кислоти (лактат) і виснаження АТФ [47]. В умовах енергодефіциту порушується функція низки трансмембранних транспортних білків, які використовують активний транспорт, зокрема K+Na+АТФази. При активації анаеробного гліколізу відбувається накопичення лактату з подальшим розвитком тяжкого внутрішньоклітинного ацидозу. Втрата клітинного гомеостазу призводить до внутрішньоклітинного накопичення натрію, кальцію. Збільшення концентрації натрію в клітині призводить до набухання і загибелі нейроцитів і гліальних елементів. Збільшення внутрішньоклітинного кальцію призводить до надмірної стимуляції рецепторів нейротрансмітерів і деполяризації мембрани [41]. Подальший приток кальцію в клітину призводить до посилення активації ліпази, що сприяє виділенню жирних кислот, збільшенню активації нейрональної NO-синтази (nNOS) й NO, що впливають на утворення вільних радикалів і дисфункції мітохондрій. Як наслідок — мітохондріальна дисфункція сигналізує про початок апоптотичної або некротичної загибелі клітин [47]. Такий механізм універсальний як для зрілого, так і незрілого головного мозку.
Проте відомо, що головний мозок, що розвивається, особливо на першому тижні життя, має більшу резистентність до гіпоксії. Цьому сприяють менша метаболічна активність незрілого мозку, його можливість активніше виводити з клітин продукти анаеробного гліколізу (лактат і кетонові тіла), а також здатність використовувати лактат і кетонові тіла як енергетичний субстрат із метою покращення оксигенації [65].
Вторинна фаза супроводжується подальшим викидом збуджуючих нейротрансмітерів і вільних радикалів, а також виснаженням запасів фосфату.
На відміну від первинної фази описані патофізіологічні феномени призводять до необоротних структурних змін у ЦНС (рис. 2Б).
Глутамат-кальцієвий стрес. У результаті енергетичного стресу відбувається масивне вивільнення збуджуючих амінокислот, зокрема глутамату, його позаклітинна концентрація значно збільшується. До того ж ситуація ускладнюється порушенням АТФ-залежного зворотного захвату глутаміну. У сірій речовині джерелом глутамату є нейрони, у білій — олігодендроцити, астроцити, а також, можливо, мікроглія. Позаклітинний глутамат активує специфічні рецептори, зокрема іонні канали NMDA- і AMPA-рецепторів. NMDA-рецептор при збудженні функціонує як кальцієвий канал, AMPA-рецептор — як натрієвий, але внаслідок незрілості субодиниці GluR2 у новонародженого (протягом перших тижнів життя) AMPA є проникним і для іонів кальцію. Тобто підвищена чутливість незрілого мозку до глутамату обумовлюється високою порівняно зі зрілим мозком експресією рецепторів NMDA і AMPA [52].
B.J. Dixon та ін. повідомляють про експресію глутаматних рецепторів у корі головного мозку, гіпокампі, базальних гангліях, таламусі, а також деяких стовбурових ядер незрілого мозку, що пояснює регіональну підвищену чутливість цих структур до перенесеної гіпоксії-ішемії [25]. Механізм глутаматопосередкованої нейрональної загибелі полягає в стійкій активації рецепторів NMDA і AMPA з порушенням натрієвого градієнта і подальшим лізисом (первинне ушкодження), а також порушенням кальцієвого градієнта, накопиченням внутрішньоклітинного кальцію і запуску кальцієвого стресу (вторинне, відтерміноване ушкодження) [51]. У результаті глутаматопосередкованої активізації кальцієвих каналів і внутрішньоклітинного енергодефіциту відбувається масивне надходження кальцію в ней-
рональні клітини. Унаслідок цього активізується низка кальцій-залежних ферментних систем, що призводить до руйнування фосфоліпідів клітинних мембран, білкових структур, ДНК і РНК, мітохондріальної дисфункції, що зрештою сприяє загибелі клітини [54].
Запальний стрес. У результаті гіпоксії-ішемії в нейрональній тканині розвивається асептичне запалення з активацією мікроглії в паренхімі мозку [53] і нейтрофілів у судинах головного мозку [56]. Незважаючи на незрілість імунної системи в новонароджених, постгіпоксична активація мікроглії і нейтрофілів у незрілому мозку спостерігається вже в перші 4–8 годин після асфіксії, що також притаманно і зрілому мозку. Наслідком цієї активації є виділення низки нейротоксичних факторів (цитокіни, оксид азоту, кисневі вільнорадикальні частинки), які потенціюють нейрональне ушкодження.
Вільнорадикальне ушкодження. В умовах гіпоксичноопосередкованого порушення мітохондріальної електронтранспортної системи та активізації запалення вивільняється велика кількість кисневих вільнорадикальних частинок, зокрема супероксиданіон (іон молекули кисню з неспареним електроном О2–). Кисневі вільнорадикальні частинки взаємодіють із молекулами-мішенями (білки, мембрани, ДНК) і руйнують чи порушують їх функції. Важливими мішенями вільнорадикального ушкодження є поліненасичені жирні кислоти й іони вільного заліза, кількість яких у незрілій нейрональній тканині вище щодо зрілої [55, 66]. У відповідь на оксидативний стрес у нейрональній тканині активується ферментна захисна система, що нейтралізує кисневі вільнорадикальні частинки: супероксиддисмутаза, каталаза і глутатіонпероксидаза. Незрілість каталази і глутатіонпероксидази та зниження їх активації призводять до підвищеної чутливості незрілого мозку до перекисного окиснення [17].
Також одним із важливих джерел кисневих вільних радикалів є активація ксантиноксидази. В умовах нормальної оксигенації ксантиноксидаза бере участь у метаболізмі пуринових основ (гіпоксантину і ксантину) з утворенням сечової кислоти, споживаючи кисень і виділяючи вільні електрони. В умовах гіпоксії така реакція інгібується з накопиченням значної кількості ксантину і гіпоксантину в клітині. При реоксигінації ксантиноксидаза різко активізується із підвищенням споживання кисню і лавиноподібним синтезом вільнорадикальних частинок.
Ще одним джерелом вільних радикалів є оксид азоту, що є важливим сигнальним агентом і нейротрансмітером. За умови гіпоксії-ішемії відбувається збільшення експресії ендотеліальної (eNOS) і нейрональної (nNOS) NO-синтази. У пізньому періоді гіпоксії-ішемії відбувається активація третьої форми (індуцибельної) NO-синтази (iNOS). У ранню фазу NO має позитивний ефект передусім за рахунок вазодилатації і покращення церебральної мікроциркуляції [36]. Негативний ефект проявляється при синтезі кисневих вільнорадикальних частинок, коли окис азоту взаємодіє із супероксиданіоном (О2–) із подальшим утворенням пероксинітриту — токсичної реактивної азотної частки (ONOO–). Пероксинітрит викликає ушкодження мембран, пригнічення цитохромоксидази і нейрональну загибель (некроз), а також сприяє активації апоптозу [19].
Активація апоптозу. Апоптоз — одна з форм програмованої загибелі клітини — виникає внаслідок активації специфічних генів і синтезу продуктів їх транскрипції. Ініціаторами апоптозу є активні форми кисню, окис азоту, глутаматкальцієвий стрес. Важливу роль у механізмі апоптозу відіграє низка ферментів, які мають назву каспази (маркери активації апоптозу). Як і некроз, апоптоз виникає практично у всіх структурах ЦНС: у кортикальних нейронах і базальних ядрах, нейронах понтосубікулярної ділянки, олігодендроцитах, нейронах мозочка, нейронах спинного мозку [46]. У низці досліджень виявлено схильність незрілої тканини до апоптозної моделі ней-рональної загибелі на противагу зрілій. При дозріванні мозку модель нейрональної загибелі зміщується від апоптозу до некрозу [68].
У низці наукових праць дослідниками виділено третій, проміжний, тип загиблих нейронів — гібридний, що в ядрах має ознаки апоптотичної загибелі, а в цитоплазмі — некротичної [24, 28, 38]. Перша згадка про некроптоз як про самостійний вид загибелі клітини було опубліковано в працях К. Тенга та співавторів у 2005 році (США) [61].
У незрілої нейрональної тканини здебільшого нейрональна загибель перебігає за комбінованим некротично-апоптозним механізмом (некроптоз). Закцентовано увагу на тому, що таке профілювання може бути пов’язане з мітохондріальною біоенергетичною недостатністю [39], що, ймовірно, призводить до припинення каскадів апоптозу при ГІУ в незрілому мозку з формуванням проміжних, гібридних типів загиблих нейронів [39, 40].
Мітохондріальна проникність. Точкою неповернення при запуску механізмів апоптозу є стан мітохондріальної проникності (англ. Mitochondrial Permeability Transition, МРТr; мітохондріальна проміжна проникність), тобто відкриття пор внутрішньої мембрани мітохондрій для проникнення молекул менш як 1500 Дальтон [42]. У мозку, який розвивається, при гіпоксії-ішемії до розвитку мітохондріальної проміжної проникності (МРТr) переважно призводять ексайтоксичність і оксидантний стрес. Вважається, що основним механізмом виникнення цих патологічних процесів є дія проапоптотичного білка X-protein (Bax) [42]. Внаслідок МРТr мітохондрії набухають і гинуть, виділяючи в цитоплазму ряд ефекторів апоптозу, а саме цитохром С, апоптозіндикуючий фактор (англ. Аpoptosis-inducing factor — AIF), прокаспазу-9 й ендонуклеазу G [42]. Цитохром С і прокаспаза-9, потрапляючи в цитоплазму, протягом 3–24 годин після ушкодження призводять до активації каспази-9, а протягом 6–48 годин — до переходу прокаспази-3 в активну каспазу-3 [28]. Активація каспази-3 забезпечує протеоліз основних клітинних білків, зокрема білків цитоскелету, а також призводить до інших морфологічних змін, які є характерними для апоптозу, зокрема фрагментації ядра [43]. Цей цитохром-опосередкований шлях також називають внутрішнім шляхом апоптозу.
Високий рівень активованої каспази-3 був виявлений у тканинах мозку померлих доношених новонароджених, які перенесли тяжку перинатальну асфіксію [44].
Існує і зовнішній шлях апоптозу, що полягає в реагуванні низки рецепторів клітинної мембрани на цитокіни при запальній стимуляції, що призводить до активації запрограмованої клітинної загибелі через активацію каспази-8 [45]. Крім цього, є також позакаспазний шлях розвитку апоптозу, що опосередкований полі(АДФ-рибоза)-полімеразою-1 (англ. Poly [ADP-ribose]-polymerase — PARP) [46]. Ефект PARP полягає в активації переходу апоптозіндикуючого фактора (AIF) із мітохондрій в ядро. Додатковим шляхом PARP є активація споживання нікотинамідаденіндинуклеотиду (Nicotinamide Adenine Dinucleotide — NAD+), необхідного для утворення мітохондріальної енергії, що сприяє вивільненню цитохрому С й активації каспази (каспазний шлях) [46].
Експериментальні дослідження показали загибель нейронів при активації PARP і зниження площі інфаркту мозку при її інгібуванні [48, 49].
Хоча кожен з описаних шляхів має свої унікальні механізми, обидва механізми «сходяться» на рівні функціонування мітохондрій, у яких змінюється проникність мембран, і в цитозоль виходять проапоптогенні фактори, зокрема апоптозіндукуючий фактор (від англ. Apoptosis inducing factor — AIF), ендонуклеаза G (endo G), цитохром С (cyt C) [60].
Отже, незважаючи на різні варіанти початку апоптозу, провідна роль належить ушкодженням на рівні мітохондрій.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Список литературы
1. Антипкін Ю.Г., Волосовець О.П., Майданник В.Г. та ін. Стан здоров’я дитячого населення — майбутнє країни (частина 2). Здоровье ребенка. 2018. Т. 13(2). С. 142-52. Режим доступу: http://nbuv.gov.ua/UJRN/Zd_2018_13_2_3.
2. Голосная Г.С., Петрухин А.С., Красильщикова Т.М. и др. Взаимодействие нейротрофических и проапоптотических факторов в патогенезе гипоксического поражения головного мозга у новорожденных. Педиатрия. 2010. Т. 89(1). С. 20-25.
3. Євтушенко С.К. и соавт. Неврология раннего детского возраста. Киев: Издательский дом «Заславский», 2016. 288 с.
4. Знаменська Т.К., Воробйова О.В., Дубініна Т.Ю. Стратегічні напрямки реконструкції системи охорони здоров’я новонароджених та дітей України. Соціальна педіатрія та реабілітологія. 2018. № 1–2(13–14). С. 7-14. Режим доступу: http://nbuv.gov.ua/UJRN/Nkhpm_2017_7_4_3.
5. Кирилова Л.Г., Мартиненко Я.А. Сучасні аспекти патогенезу ураження головного мозку в дітей, котрі народилися з екстремально низькою масою тіла. Перинатологія і педіатрія. 2015. № 4. С. 64-68. Режим доступу: http://nbuv.gov.ua/UJRN/perynatology_2015_4_14.
6. Мартинюк В.Ю. Метаболічна енцефалопатія, як один з факторів, що обумовлюють формування інвалідізації у дітей. Український медичний вісник психоневрології. Харків, 1995. Т. ІІІ. 2(6). С. 363-365.
7. Моісеєнко Р.О., Гойда Н.Г., Дудіна О.О. Дитяча інвалідність та питання розбудови системи медико-соціальної реабілітації дітей в Україні. Соціальна педіатрія та реабілітологія. 2018. № 3–4 (15-16). С. 10-19.
8. Пальчик А.Б., Шабалов Н.П. Гипоксически-ишемическая энцефалопатия новорожденных. 4-е изд., испр. и доп. Москва: МЕДпресс-информ, 2013. 288 с.
9. Семенова К.А. Восстановительное лечение детей с перинатальными поражениями нервной системы и детским церебральным параличом. Москва: Закон и порядок, 2007. 616 с.
10. Abukahma A.F., Mousa A.Y., Stone P.A. Shunting during carotid endarterectormy. J. Vasc. Surg. 2011. Nov. 54(5). 1502-1510. DOI: 10.1016/j.jvs.2011.06.020. PMID: 21906905.
11. Allen K.A., Brandon D.H. Hypoxic ischemic encephalopathy: pathophysiology and experimental treatments. Newborn Infant Nurs. Rev. 2011. Sep 1. 11(3). 125-133. DOI: 10.1053/j.nainr.2011.07.004. PMCID: PMC3171747. NIHMSID: NIHMS310162. PMID: 21927583.
12. Auten R.L., Dovits J.M. Oxygen toxicity and reactive oxygen specils:the devil is in the details. Pediatr. Res. 2009 Aug. 66(2). 121-127. DOI: 10.1203/PDR.0b013e3181a9eafb. PMID: 19390491.
13. Azzopardi D., Wyatt J.S., Cady E.B. et al. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989. 25(5). 445-451. DOI: 10.1203/00006450-198905000-00004. PMID: 2717259.
14. Bano S., Chaudhary V., Garga U. C. Neonatal Hypoxic-ischemic Encephalopathy: A Radiological Review. J. Pediatr. Neurosci. 2017. Jan-Mar. 12(1). 1-6. DOI: 10.4103/1817-1745.205646. PMID: 28553370. PMCID: PMC5437770.
15. Baptiste-Roberts К., Salafia C.M., Nicholson W.K., Duggan A., Wang N.Y., Brancati F.L. Maternal risk factors for abnormal placental growth: the national collaborative perinatal project. BMC Pregnancy Childbirth. 2008. Sep. 8. 44. DOI: 10.1186/1471-2393-8-44. PMID: 18811957. PMCID: PMC2564930.
16. D. L. Neurotrophic factors: important regulators of nociceptive function. Neuroscientist. 2000. 7(1). 13-17. DOI: 10.1177/107385840100700105. PMID: 11486340.
17. Blomgren K., Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006. Feb 1.40(3). 388-397. DOI: 10.1016/j.freeradbiomed.2005.08.040. PMID: 16443153.
18. Borovski D., Czuba B., Włoch A., Czuba B., Włoch A. еt аl. Doppler assessment of the fetal asphyxia in pregnancies complicated by gestational hypertension and intrauterine gronth retardation. Gynecol. Pol. 2006. Mar. 77(3). 184-189. PMID: 16871835.
19. Calcerrada P., Peluffo G., Radi R. Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications. Curr. Pharm. Des. 2011. Dec. 17(35). 3905-3932. DOI: 10.2174/138161211798357719. PMID: 21933142.
20. Chalak L.F. Inflammatory Biomarkers of Birth Asphyxia. Clin. Perinatol. 2016. Sep. 43(3). 501-510. DOI: 10.1016/j.clp.2016.04.008. PMID: 27524450. PMCID: PMC6170165.
21. Chaparro-Huerta V., Flores-Soto M.E., Merin Sigala M.E. et al. Proinflammatory Cytokines, Enolase and S-100 as Early Biochemical Indicators of Hypoxic-Ischemic Encephalopathy Following Perinatal Asphyxia in Newborns. Pediatr. Neonatol. 2017. Feb. 58(1). 70-76. DOI: 10.1016/j.pedneo.2016.05.001. PMID: 27522459.
22. Cotten C.M., Shankaran S. Hypothermia for hypoxic-ischemic encephalopathy. Expert Rev. Obstet. Gynecol. 2010. 5(2). 227-239. DOI: 10.1586/eog.10.7.
23. De Menezes M. S. Hypoxic-ischemic brain injury in the newborn. 2006. URL: http://emedicine.medscape.com/article/1183351 (дата обращения: 20.05.2011).
24. Deev R.V., Bilyalov A.I., Zhampeisov T.M. Modern ideas about cell death. Genes & Cells. 2018. XIII(1). 6-19. DOI: 10.23868/201805001.
25. Dixon B.J., Reis C., Ho W.M. et al. Neuroprotective strategies after neonatal hypoxic ischemic encephalopathy. Int. J. Mol. Sci. 2015. Sep. 16(9). 22368-22401. DOI: 10.3390/ijms160922368. PMID: 26389893. PMCID: PMC4613313.
26. Douglas-Escobar M., Weiss M.D. Hypoxic-ischemic encephalopathy: а review for the clinician. JAMA Pediatr. 2015. Apr. 169(4). 397-403. DOI: 10.1001/jamape diatrics.2014.3269. PMID: 25685948.
27. Eslamian L., Tooba K. Doppler findings in intrapartum fetal distress. Acta Med. Iran. 2011. 49(8). 547-550. PMID: 22009812.
28. Esposito E., Cuzzocrea S. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010. Sep. 8(3). 228-242. DOI: 10.2174/157015910792246155. PMID: 21358973. PMCID: PMC3001216.
29. Ferriero D.M. Neonatal brain injury. N. Engl. J. Med. 2004. Nov. 4. 351(19). 1985-95. DOI: 10.1056/NEJMra041996. PMID: 15525724.
30. Fleiss B., Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy. Lancet Neurol. 2012. 11(6). 556-566. DOI: 10.1016/s1474-4422(12)70058-3.
31. Gerner G.J., Burton V.J., Poretti A. et al. Transfontanellar duplex brain ultrasonography resistive indices as a prognostic tool in neonatal hypoxic-ischemic encephalopathy before and after treatment with therapeutic hypothermia. J. Perinatol. 2016. Mar. 36(3). 202-206. DOI: 10.1038/jp.2015.169. PMID: 26609871. PMCID: PMC4767581.
32. Graham E.M., Burd I., Everett A.D., Northington F.J. Blood Biomarkers for Evaluation of Perinatal Encephalopathy. Front. Pharmacol. 2016. Jul. 13(7). 196. DOI: 10.3389/fphar.2016.00196. PMID: 27468268. PMCID: PMC4942457.
33. Grow J., Barks J. D. Pathogenesis of hypoxic-ischemic cerebral injury in the term infant: current concepts. Clin. Perinatol. 2002. Dec. 29(4). 585-602. DOI: 10.1016/s0095-5108(02)00059-3. PMID: 12516737.
34. Hassell K.J., Ezzati M., Alonso-Alconada D. et al. New horizons for newborn brain protection: enhancing endogenous neuroprotection. Arch. Dis. Child Fetal. Neonatal Ed. 2015. 100(6). 541-552. DOI: 10.1136/archdischild-2014-306284.
35. Hill C.A., Fitch R.H. Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: implications for sex-specific neuroprotection in clinical neonatal practice. Neurol. Res. Int. 2012. 2012. 867531. DOI: 10.1155/2012/867531. PMID: 22474588. PMCID: PMC3306914.
36. Ioroi T., Yonetani M., Nakamura H. Effects of hypoxia and reoxygenation on nitric oxide production and cerebral blood flow in developing rat striatum. Pediatr. Res. 1998. Jun. 43(6). 733-737. DOI: 10.1203/00006450-199806000-00004. PMID: 9621981.
37. Iwata O., Iwata S., Thornton J.S. et al. Therapeutic time window duration decreases with increasing severity of cerebral hypoxia-ischaemia under normothermia and delayed hypothermia in newborn piglets. Brain Res. 2007. Jun. 18(1154). 173-180. DOI: 10.1016/j.brainres.2007.03.083. PMID: 17475224.
38. Jacobs S.E., Berg M., Hunt R., Tarnow-Mordi W.O., Inder T.E., Davis P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013. Jan. 31(1). CD003311. DOI: 10.1002/14651858.CD003311.pub3. PMID: 23440789. PMCID: PMC7003568.
39. Jandova K., Riljak V., Maresova D. et al. Ascorbic acid and alpha-tocopherol protect age-dependently from hypoxia-induced changes of cortical excitability in developing rats. Neuro Endocrinol. Lett. 2012. 33(5). 530-535. PMID: 23090272.
40. Jou M.J., Peng T.I., Yu P.Z. еt al. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal. Res. 2007. Nov. 43(4). 389-403. DOI: 10.1111/j.1600-079X.2007.00490.x. PMID: 17910608.
41. Juul S.E., Ferriero D.M. Pharmacologic neuroprotective strategies in neonatal brain injury. Clin. Perinatol. 2014. 41. 119-131. DOI: 10.1016/j.clp.2013.09.004. [PMC free article] [PubMed] [CrossRef] [Google Scholar].
42. Kaandorp J.J., Benders M.J., Schuit E. et al. Maternal allopurinol administration during suspected fetal hypoxia: a novel neuroprotective intervention? A multicentrerandomised placebo controlled trial. Arch. Dis. Child Fetal. Neonatal. Ed. 2015. May. 100(3). 216-223. DOI: 10.1136/archdischild-2014-306769. PMID: 25512466.
43. Kawakami M., Sekiguchi M., Sato K., Kozaki S., Takahashi M. Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J. Biol. Chem. 2001. Oct. 276(42). 39469-39475. DOI: 10.1074/jbc.M105832200. PMID: 11504731.
44. Koh S., Tibayan F.D., Simpson J.N., Jensen F.E. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004. Jun. 45(6). 569-575. DOI: 10.1111/j.0013-9580.2004.69103.x. PMID: 15144420. PMID: 15144420.
45. Kohmura E., Yamada K., Hayakawa T., Kinoshita А. Neurotoxicity caused by glutamate after subcritical hypoxia is prevented by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX): an in vitro study using rat hippocampal neurons. Neurosci. Lett. 1991. Jan. 121(1–2). 159-162. https://DOI.org/10.1016/0304-3940(91)90674-I.
46. Kumral A., Gonenc S., Acikgoz O. et al. Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxicischemic brain injury in neonatal rats. Biol. Neonate. 2005. 87(1). 15-18. DOI: 10.1159/000080490. PMID: 15334031.
47. Lai M.C., Yang S.N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011. 609813. DOI: 10.1155/2011/609813. PMID: 21197402. PMCID: PMC3010686.
48. Lapchak P.A., Zivin J.A. Ebselen, a seleno-organic antioxidant, is neuroprotective after embolic strokes in rabbits: synergism with low-dose tissue plasminogen activator. Stroke. 2003. Aug. 34(8). 2013-2018. DOI: 10.1161/01.STR.0000081223.74129.04. PMID: 12855833.
49. Lin C.Y., Tsai P.S., Hung Y.C. et al. L-type calcium channels are involved in mediating the anti-inflammatory effects of magnesium sulphate. Br. J. Anaesth. 2010. Jan. 104(1). 44-51. DOI: 10.1093/bja/aep336. PMID: 19933511.
50. Martyniuk V. Critical condition as a nonspecific causal factor of developmental disorders of nervous system. European Journal of Paediatric Neurology. 1999. 3. ISSUE 6. 40. DOI: 10.1016/S1090-3798(99)91091-7.
51. Matute C., Alberdi E., Domercq M. еt al. Excitotoxic da-mage to white matter. J. Anat. 2007. Jun. 210(6). 693-702. DOI: 10.1111/j.1469-7580.2007.00733.x. PMCID: PMC2375761. PMID: 17504270.
52. McQuillen P.S., Ferriero D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004. Apr. 30(4). 227-235. DOI: 10.1016/j.pediatrneurol.2003.10.001. PMID: 15087099.
53. McRae A., Gilland E., Bona E. еt al. Microglia activation after neonatal hypoxic-ischemia. Brain. Res. Dev. Brain. Res. 1995. Feb. 84(2). 245-252. DOI: 10.1016/0165-3806(94)00177-2. PMID: 7743644.
54. Morley P., Hogan M. J., Hakim A. M. Calcium-mediated mechanisms of ischemic injury and protection. Brain Pathol. 1994. 4(1). 37-47. DOI: 10.1111/j.1750-3639.1994.tb00809.x. PMID: 8025702.
55. Ogihara T., Hirano K., Ogihara H. еt al. Non-protein-bound transition metals and hydroxyl radical generation in cerebrospinal fluid of newborn infants with hypoxic ischemic encephalopathy. Pediatr. Res. 2003. 53(4). 594-599. DOI: 10.1203/01.PDR.0000054685.87405.59.
56. Palmer C., Roberts R.L., Young P.I. Timing of neutrophil depletion influences long-term neuroprotection in neonatal rat hypoxic-ischemic brain injury. Pediatr. Res. 2004. 55(4). 549-556. DOI: 10.1203/01.PDR.0000113546.03897.FC. PMID: 14739365.
57. Perlman J. M. Brain injury in the term infant. Semin. Perinatol. 2004. 28(6). 415-424. DOI: 10.1053/j.semperi.2004.10.003.
58. Rabinstein A., Resnick S. Hypoxic-ischemic brain damage. Practical Neuroimaging in Stroke: A CaseBased Approach. Medical. 2009. Vol. 1. 1-17.
59. Risso F.M., Sannia A., Gavilanes D.A. et al. Biomarkers of brain damage in preterm infants J. Matern. Fetal. Neonatal. Med. 2012. 25(54). 101-104. DOI: 10.3109/14767058.2012.715024.
60. Rousset C.I., Baburamani A.A., Thornton C. et al. Mitochondria and perinatal brain injury. J. Matern. Fetal. Neonatal. Med. 2012. Suppl. 1.35-38. DOI: 10.3109/14767058.2012.666398. PMID: 22348594.
61. Teng X.M., Degterev A., Jagtap P. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett. 2005. Nov. 15(22). 5039-44. DOI: 10.1016/j.bmcl.2005.07.077. PMID: 16153840.
62. Vaux D. Apoptosis Timeline. Cell Death & Differentiation. 2002. 9. 349-354. DOI: 10.1038/sj.cdd.4400990.
63. Volpe J.J. Neurology of the newborn. 5th. Saunders Elsevier, 2008. 1120 р.
64. Wang K.K., Yang Z., Sarkis G., Torres I., Raghavan V. Ubiquitin C-terminal hydrolase-L1 (UCH-L1) as a therapeutic and diagnostic target in neurodegeneration, neurotrauma and neuro-injuries.Expert Opin. Ther. Targets. 2017. Jun. 21(6). 627-638. DOI: 10.1080/14728222.2017.1321635. PMID: 28434268.
65. Yager J.Y., Thornhill J.A. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci. Biobehav. Rev. 1997. Mar. 21(2). 167-74. DOI: 10.1016/s0149-7634(96)00006-1. PMID: 9062939.
66. Yu T., Kui L.Q., Ming Q.Z. Effect of asphyxia on non-protein-bound iron and lipid peroxidation in newborn infants. Dev. Med. Child Neurol. 2003. Jan. 45(1). 24-27. PMID: 12549751.
67. Zanelli S.A, Stanley D. P., Kaufman D. Hypoxic-ischemic encephalopathy. 2012. URL: http://emedicine.medscape.com/article/973501.
68. Zhu C., Wang X., Xu F. et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell. Death Differ. 2005. Feb. 12(2). 162-76. DOI: 10.1038/sj.cdd.4401545. PMID: 15592434.
Продовження статті
в наступному номері

/13.jpg)
/15.jpg)