Наявність у пацієнта декількох ендокринних захворювань, особливо коли йдеться про надлишок гормонів, завжди має викликати в лікаря підозру щодо синдромів множинної ендокринної неоплазії та інших генетичних синдромів, які викликають поліорганні пухлини.
Клінічний випадок
Жінка 39 років звернулася за консультацією з клінічними ознаками гіперкортицизму (місяцеподібне обличчя, абдомінальне ожиріння, артеріальна гіпертензія і вторинна аменорея). Лабораторно був підтверджений синдром Кушинга АКТГ (адренокортикотропний гормон)-незалежний, проте виконана за місцем проживання комп’ютерна томографія не виявила утворень у надниркових залозах.
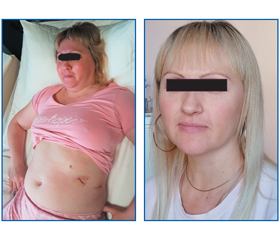
Установлення причини низького рівня АКТГ ускладнював анамнез пацієнтки, адже 2 роки тому вона перенесла видалення аденоми гіпофіза (соматотропіноми). З огляду на проведену діагностику акромегалії в здійснених лабораторних дослідженнях вже визначався знижений АКТГ, проте з урахуванням нормальних рівнів кортизолу в крові (дексаметазоновий тест не проводили) на це не звернули увагу. У той час причинами звернення пацієнтки були набір маси тіла, дисменорея, випадіння волосся та зміни зовнішності, які продовжувалися навіть після видалення макроаденоми гіпофіза (рис. 1, 2: фото пацієнтки; рис. 3: МРТ гіпофіза до операції).
Серед інших проблем зі здоров’ям пацієнтка відзначала мігрені впродовж останніх 15 років, дисменорею останні 3 роки, наявність двох вузлів у матці і двох вузлів у щитоподібній залозі. В анамнезі також дві нормальні вагітності (1998 і 2012 роки). Крім того, серед встановлених діагнозів звертала на себе увагу кардіоміопатія складного генезу — дифузні зміни в міокарді і синусова брадикардія. Сімейний анамнез обтяжений гострим лейкозом у батька і тромбом, що призвів до смерті, в бабусі.
Перегляд попередньо проведених лабораторних аналізів призвів до думки про АКТГ-незалежний синдром Кушинга, зумовлений первинною пігментною вузликовою гіперплазією кори надниркових залоз (PPNAD — primary pigmented nodular adrenal disease). Особливу увагу викликало пародоксальне підвищення кортизолу в сечі після проведення дводенної малої дексаметазонової проби (рівень кортизолу в сечі збільшився з 298,54 до 423,45 мкг/24 години при нормальних значеннях 50–190 мкг/24 год), а також зниження рівня дегідроепіандростерону сульфату (28,2 і 26,6 мкг/дл при нормальних рівнях 60,9–337,0 мкг/дл для її віку).
З огляду на анамнез був запідозрений комплекс Карні (КК), пацієнтці було рекомендовано пройти ехокардіографію для виключення міксоми серця. Дане дослідження дійсно виявило міксому перетинки серця до 10 мм у діаметрі (рис. 4: міксома серця).
Пацієнтці було виконано дві комп’ютерні томографії надниркових залоз, які не виявили істотних додаткових утворень у них (рис. 5: КТ надниркових залоз, фронтальний зріз). Водночас, враховуючи динаміку на знімках КТ надниркових залоз, а також підозру на PPNAD у даної хворої, прийняли рішення про хірургічне (лапароскопічне) видалення більшої (лівої) залози (рис. 6, 7: КТ надниркових залоз — більш виражена гіперплазія лівого). Лапароскопічний вигляд зміненої внаслідок PPNAD надниркової залози був дуже показовим (рис. 8: лапароскопічний вигляд надниркової залози через тонкий шар очеревини), а макроскопічна картина (рис. 9: препарат видаленої лівої надниркової залози з типовими для PPNAD змінами) та гістологічний висновок підтвердили припущення щодо природи гіперкортизолемії в пацієнтки.
/103.jpg)
У післяопераційному періоді замісна гормональна терапія не була призначена, адже залишалася друга залоза, уражена цією ж хворобою, і рівень кортизолу в крові після операції очікувано мав бути принаймні нормальним або підвищеним. Результат виміру кортизолу в крові на другу добу після операції показав нижньонормальне значення — 130 нмоль/мл (норма — 126–600).
Через тиждень після операції неочікувано виникли симптоми надниркової недостатності (кортизол сечі був 60 мкг/л/24 год, норма — 60–340) і була призначена замісна терапія гідрокортизоном, на якій пацієнтка залишається вже більше 10 місяців. Спроби знизити дозування з 10 до 5 мг гідрокортизону призводили до повернення симптомів гіпокортицизму. Загалом стан пацієнтки швидко покращився — відновились місячні, повернулась до нормальної маса тіла та зовнішність (рис. 10: вигляд пацієнтки на другу добу після лапароскопічної адреналектомії; рис. 11: зовнішність хворої через 6 міс. після адреналектомії).
Обговорення
Комплекс Карні (КК) — рідкісний синдром множинної неоплазії (MIM 160980), що успадковується за автосомно-домінантним типом. Цю хворобу можна характеризувати наявністю пігментних утворень шкіри або слизової, міксомами серця, шкіри та інших органів, а також множинними ендокринними та неендокринними пухлинними утвореннями. За своїми характеристиками і різноманіттям цей синдром схожий на нейрофіброматоз, факоматоз, гамартроз, синдром Мак’юна — Олбрайта, Пейтца — Єгерса, Ковдена та інші. Вперше описаний у 1985 році Дж.Е. Карні в клініці Мейо як наявність міксом, гіперпігментацї шкіри та підвищення активності гормонів [1].
Поширеність хвороби невідома, але дуже низька. У літературі описано приблизно 750 випадків, з яких 70 % — родинні.
Клінічні прояви комплексу Карні дуже різноманітні, і повна картина може розвиватися через багато років. Інколи діагноз установлюють після 40 років, у середньому — у віці 20 років.
Ураження шкіри
Ураження шкіри виникає в більше ніж 80 % пацієнтів і є одним із найяскравіших проявів цієї хвороби: від пігментних плям, лентіго, блакитних невусів до міксом шкіри.
Лентіго (пласкі, маленькі коричневі або чорні макули) виникають зазвичай до пубертату, після якого може збільшитися їх кількість й інтенсивність забарвлення. Вони можуть бути розташовані по всьому тілу, але з переважанням у зоні обличчя, на губах, слизових і геніталіях. Їх яскравість може зменшуватися з віком.
Епітеліоїдний блакитний невус — куполоподібна папула синюватого відтінку з гладенькою поверхнею — рідкісний субтип блакитного невусу в загальній популяції, проте відносно часто трапляється серед пацієнтів із комплексом Карні. Останнє має стимулювати лікарів до можливого пошуку цього синдрому серед таких хворих. Епітеліоїдний блакитний невус може бути інтенсивно забарвлений і погано окреслений, з асоційованим дермальним фіброзом.
Шкірні міксоми виявляють у 30–55 % випадків КК, із найчастішою локалізацією на повіках, зовнішньому вушному каналі, сосках і геніталіях. Зазвичай вони до 1 см у діаметрі, безболісні, розташовані в дермі або підшкірно.
Серцеві прояви
Найбільш поширеними нешкірними утвореннями в комплексі Карні є міксоми серця. Вони спостерігаються у 20–40 % пацієнтів і найчастіше виявляються у віці 20 років, хоча можуть бути діагностовані й у дитинстві. Міксоми серця можуть уражувати будь-який його відділ і трапляються з однаковою частотою в жінок і чоловіків на відміну від спорадичних міксом, які уражують в основному ліве передсердя в жінок старшого віку. Проявами хвороби є ознаки внутрішньосерцевої обструкції або емболії з подальшими інсультами та ознаками серцевої недостатності, що є причиною смерті в 50 % комплексу Карні.
Міксоми можуть повністю перекривати клапанний отвір і ставати причиною раптової смерті. Тому особлива увага має приділятися саме ранній діагностиці, для якої ефективною буде звичайна ехокардіографія (також інформативними будуть комп’ютерна томографія та магнітно-резонансна томографія, особливо в післяопераційних випадках, коли анатомія серця порушена) [2].
В одному з останніх досліджень із Китаю частота міксом за комплексу Карні становила лише 1,74 %, але рівень рецидивів статистично перевищував такий при спорадичних міксомах. В описаних випадках спостерігалася суттєва мітральна і помірна трикуспідальна регургітація [3].
Ураження гіпофіза
Підвищення гормона росту, інсуліноподібного фактора росту 1-го типу (ІФР-1) та пролактину трапляється майже в 75 % випадків, навіть без явних утворень у гіпофізі. Поширеність акромегалії в пацієнтів із комплексом Карні становить 10–12 %. У поодиноких випадках були описані пролактиноми у пацієнтів із комплексом Карні. Аденома зазвичай виникає на другій-третій декадах життя. Гістологічно звертає на себе увагу гіперплазія соматоморфних клітин, що може пояснювати поступовий початок хвороби. Лікування проводять аналогами соматостатину або хірургічним видаленням аденоми гіпофіза [2, 4].
У найбільшому огляді описано 57 випадків акромегалії в пацієнтів із КК (24 чоловіки і 33 жінки), середній вік на момент установлення діагнозу становив 28 ± 12 років (для спорадичної акромегалії — 40–48 років). Приблизно половину випадків становили мікроаденоми, тоді як в інших випадках акромегалії переважають макроаденоми [5].
Ураження надниркових залоз
Відокремлення комплексу Карні в особливий симптомокомплекс відбулося після вивчення одного з видів АКТГ-незалежного синдрому Кушинга — первинної пігментної вузликової гіперплазії кори надниркових залоз (PPNAD). Назва цього виду гіперкортизолемії походить від макроскопічного вигляду надниркових залоз — множинних пігментованих вузликів, що уражують кортикальний шар обох надниркових залоз. PPNAD діагностують у складі комплексу Карні у 25–60 % випадків, проте бувають й ізольовані форми. Ця гіперплазія частіше виникає в дітей і дорослих віком до 40 років. Діагностика часто ускладнюється тривалим помірним перебігом хвороби, хоча описані випадки і різкого прогресування гіперкортизолемії, а також циклічним синдромом Кушинга. Середній час від появи перших проявів хвороби до встановлення діагнозу становить 4 роки [2, 4, 6].
Синдром Кушинга за даної патології не відрізняється нічим особливим, основними симптомами є місяцеподібне обличчя й абдомінальне ожиріння. Стрії, екхімози і м’язова слабкість також будуть класичними ознаками. Ураховуючи високий відсоток дітей у цій групі, треба зважати і на зміни кривих росту, проте класична затримка росту спостерігається не так часто, як в інших випадках гіперкортизолемії (до 30 %) [2, 7]. Останнє можна пояснити циклічною секрецією гормонів у певних випадках. Остеопороз трапляється достатньо часто, особливо за умови пізньої діагностики.
Вільний кортизол у сечі підвищений у більшості пацієнтів, а добова циклічність секреції кортизолу порушена. Як і для інших причин синдрому Кушинга надниркового генезу, рівень АКТГ знижений, хоча в деяких випадках визначається нижня межа нормальних значень. Усі тести з пригніченням дексаметазоном (нічний, малий і великий) будуть вказувати на автономну гіперсекрецію кортизолу. Також характерна парадоксальна реакція кортизолу в сечі під час проведення великої дексаметазонової проби (8 мг) із збільшенням рівня кортизолурії майже в 100 % випадків.
Морфологічно кора надниркових залоз усіяна дрібними пігментованими, темними вузликами до 5–10 мм. Патогістологічне дослідження надниркових залоз зазвичай виявляє їх нормальні розміри (вага 4–17 г). Стає зрозумілим, що для радіологічної діагностики треба використовувати комп’ютерну томографію з товщиною зрізу до 3 мм, адже лише тоді можна виявити чітко окреслені, округлі утворення зниженої щільності. Варто зауважити, що за умови тривалого перебігу хвороби ці вузлики можуть зростати понад 10 мм.
Лікування — хірургічне, що полягає в лапароскопічному видаленні обох надниркових залоз із подальшим призначенням замісної терапії [2, 4].
Ураження щитоподібної залози
Пацієнти з комплексом Карні мають утворення в щитоподібній залозі майже в 60 % випадків. Переважають кістозні утворення (до 75 %) і доброякісні вузли (до 25 %), до 10 % випадків діагностують папілярний або фолікулярний рак щитоподібної залози. Часто вузлові утворення виникають ще в перші 10 років життя. Порушення функції майже не відзначаються. З огляду на ці дані пункційна біопсія є обов’язковою в більшості випадків утворень у ЩЗ із подальшим лікуванням за відповідними світовими протоколами [2, 4].
Ураження нервової системи
Пігментна псаммоматозна шваннома — рідкісна пухлина нервових оболонок, що описана в пацієнтів із КК (до 10 %). Вона може бути розташована в будь-якій ділянці периферичної або центральної нервової системи, часто мультицентрична, пігментна і містить кальцифікати. Описані випадки розташування в стравоході, шлунку, печінці, прямій кишці, грудній стінці і параспінально. Пухлини проявляються болем і радикулопатією та можуть мати ризик злоякісності. Лікування зазвичай хірургічне, що ускладнюється розташуванням цих пухлин [2, 4].
Офтальмологічні прояви
Найчастішими офтальмологічними симптомами КК є пігментні утворення карункулу чи напівмісячної складки, лентиго і міксоми повік. Іноді описують можливість пігментних шванном райдужної оболонки.
Ураження яєчок
Більше половини чоловіків із КК мають великоклітинні кальцинуючі пухлини з клітин Сертолі. Ці утворення зазвичай пальпуються, можуть бути білатеральними, мультифокальними, мають певні ризики малігнізації та зниження фертильності. Основним методом діагностики буде ультразвукове дослідження. У хлопців та юнаків можуть виникнути гінекомастія, дострокове закриття зон росту та раннє статеве дозрівання, що може потребувати хірургічного лікування та навіть призначення інгібіторів ароматаз. Інші види утворень зустрічаються набагато рідше [2, 4].
Інші ураження
Міксоми молочних залоз і сосків достатньо поширені в будь-якому віці і навіть у чоловіків. Щодо яєчників описані різні види патології — від кіст і тератом до раку яєчників. Остеохондроміксома є рідкісним компонентом комплексу Карні, проте описані випадки навіть у дитячому віці. Частіше уражуються назальні синуси, а також трубчасті кістки кінцівок. Мають доброякісний характер, однак схильні до рецидивування. Гепатоцелюлярні аденоми, міксоми матки, пухлини підшлункової залози, бронхів, прищитоподібних залоз, шлунка, товстого кишечника та інші пухлини також були описані з низькою поширеністю [2, 4].
Діагностика
Діагноз комплексу Карні слід запідозрити за фенотиповими ознаками з подальшим генетичним підтвердженням, що, на жаль, поки що в Україні недоступне. Середній вік на момент установлення діагнозу у світі — 20 років.
Без генетичного підтвердження діагноз можна встановити на підставі наявності певних критеріїв (табл. 1). Для встановлення діагнозу КК потрібні 2 основні критерії або 1 основний й один додатковий. Малі критерії можуть асоціюватися з КК, але не є діагностичними [2, 4].
Генетика
КК спричинений мутацією гена PRKAR1A (ОМІМ 188830) у 70–80 % випадків. Він кодує регуляторну субодиницю типу 1-альфа ферменту протеїнкінази А. Цей ген розташований у 24.2–24.3 локусах довгого плеча 17-ї хромосоми і має 11 екзонів. Також нещодавно були описані мутації в генах PRKAСА та PRKAСВ.
Лікування
Кожна специфічна медична проблема в КК потребує свого конкретного вирішення і звернення до відповідних вузьких спеціалістів — часто з подальшим хірургічним лікуванням.
Спостереження
Адекватне спостереження значно покращує загальний прогноз. Отже, має проводитися з принаймні річною регулярністю:
1) щорічна ехокардіографія, починаючи з раннього віку;
2) регулярні огляди в дерматолога;
3) аналіз крові на гормон росту, ІФР-1, пролактин й оцінка рівня кортизолу (нічний дексаметазоновий тест, вільний кортизол добової сечі або нічний кортизол слини) із підліткового віку;
4) клінічне обстеження щитоподібної залози;
5) щорічне ультразвукове обстеження яєчок та яєчників;
6) у дітей до пубертату щорічна оцінка швидкості росту і стадії статевого дозрівання.
Прогноз
Описана тривалість життя пацієнтів із КК становить 50–55 років, проте за умови адекватного спостереження та лікування може бути набагато більшою. Основною причиною смерті стають зазвичай серцево-судинні ускладнення міксом та наслідки їх лікування (тромбози, аритмії, кардіоміопатії та інсульти), а також онкологічні захворювання.
З урахуванням усього наведеного вище, навіть без проведеного генетичного аналізу, стає очевидним наявність комплексу Карні в нашої пацієнтки. Підозра на це захворювання дала змогу виявити міксому серця, що надасть можливість безпечного спостереження за нею в подальшому. Крім того, діти пацієнтки будуть обстежені і знатимуть про можливі ризики щодо здоров’я в майбутньому. Вкрай позитивна динаміка синдрому Кушинга після видалення однієї з надниркових залоз попри міжнародні рекомендації щодо одномоментного видалення обох залоз свідчить на користь більш поміркованого ставлення до тотальної адреналектомії з приводу PPNAD у цієї групи хворих. Остання теза потребує подальших досліджень.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.

/103.jpg)
/104.jpg)
/106.jpg)
/107.jpg)