Краткая характеристика интерлейкина-17
Провоспалительный цитокин интерлейкин-17 (ИЛ-17) является Т-клеточным полипептидом, секретируемым преимущественно субпопуляцией CD4+ Т-хелперов (Th17-клетками), а также ЕК-клетками, нейтрофилами, макрофагами, дендритными, плазматическими и тучными клетками. ИЛ-17 впервые был описан в 1953 г., а как уникальная Т-хелперная клеточная линия идентифицирован в 2005 г. двумя группами авторов [1, 2]. Первоначальное название ИЛ-17 — CTLA.
Семейство ИЛ-17 состоит из 6 членов: ИЛ-17А, ИЛ-17В, ИЛ-17G, ИЛ-17D, ИЛ-17E (ИЛ25) и ИЛ-17F. Молекулярная масса ИЛ-17А — 15,1 кДа, ИЛ-17F — 14,9 кДа. Гены ИЛ-17А и ИЛ-17F локализованы на длинном плече хромосомы G (Gq12) [3]. Из всех членов семейства ИЛ-17 наиболее активным и изученным является ИЛ-17А, поэтому в большинстве публикаций он обозначается собирательным термином ИЛ-17. Основными мишенями ИЛ-17 являются лейкоциты, эпителиальные и эндотелиальные клетки, а также фибробласты, на цитоплазматической мембране которых имеются специфические к нему рецепторы (ИЛ-17Р), включающие также другие рецепторы семейства ИЛ-17 (ИЛ-17А, ИЛ-17В, ИЛ-17С, ИЛ-17D и ИЛ-17Е) [4].
В настоящее время ИЛ-17 считают одним из важнейших регуляторов естественного и адаптивного иммунитета в организме, особенно проявляющегося при различных воспалительных заболеваниях и аутоиммунных нозологиях, включающих сахарный диабет, а также онкологические заболевания.
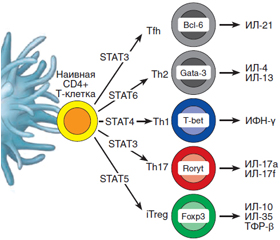
Как известно, Т-хелперы (CD4+ Т-клетки) являются клетками-«помощниками», которые занимают центральное место в регуляции иммунитета и в то же время способствуют экспансии и потенцируют функцию цитотоксических СD8+ Т-клеток и продукцию антител В-лимфоцитами. Среди молодых периферических CD4+ T-клеток различают два субкласса: наивные клетки и клетки памяти. Согласно общепринятой точке зрения, Th17-клетки, секретирующие ИЛ-17, образуются из наивных CD4+ Т-хелперов после их контакта с антигенпрезентирующими дендритными клетками. После этого наивные CD4+ T-клетки, как видно из рис. 1, способны дифференцироваться в Th1-, Th2-, Th9-, регуляторные (CD4+CD25+FoxP3+) и фолликулярные хелперные (Tfh) клетки в зависимости от воздействия специфических цитокиновых сигналов и различных транскрипционных факторов STAT 1-6 (Signal Transducer and Activator of Transcription) [5].
/93-1.jpg)
В образовании, пролиферации и функциональной активности Th17-клеток ключевую роль играют транскрипционный фактор STAT3, а также цитокины: трансформирующий фактор роста бета (ТФР-β), ИЛ-6, ИЛ-21 и ИЛ-23.
Кроме того, Th17-клетки характеризуются экспрессией транскрипционного фактора Retinoic acid Receptor, связанного Organ Receptor гамма тимуса (RORγt у мыши и RORC у человека) и способностью секретировать семейство ИЛ-17 и цитокины ИЛ-22, ИЛ-23Р, GM-CSF, хемокин CCR-рецептор и потенциально ИЛ-6 и ФНО-α. Причем ТФР-β и ИЛ-23 могут присутствовать локально в микроокружении. Особенностью Th17 являются также большая пластичность и способность к поляризации, т.е. возможность дифференцироваться в другие виды клеток, в том числе одновременно содержать FoxP3 и RORγt [4, 6, 7].
Механизм последующей дифференциации ИЛ-17 весьма сложен и изучается в основном на молекулярном уровне с использованием преимущественно модели спонтанного аутоиммунного диабета животных (в основном NOD-мышей) [8]. Вместе с тем следует отметить, что невозможно полностью экстраполировать иммунологические данные, полученные на лабораторных животных, на иммунные реакции, специфически протекающие только у человека, в частности, страдающего сахарным диабетом (СД), ввиду их различных видовых и социальных особенностей [6, 9].
Процесс дифференцировки Th17-клеток может быть условно разделен на три стадии: 1) дифференцировка под воздействием ИЛ-6 и ТФР-β, которая приводит к трансформации наивных CD4+ Т-клеток в Th17-клетки при участии транскрипционных факторов STAT3 и RORγt (у мыши) или RORC (у человека); затем ТФР-β способствует повышению восприимчивой чувствительности наивных Т-клеток к ИЛ-23, увеличивая экспрессию их рецептора; 2) стадия стабилизации и экспансии Th1-клеток под влиянием ИЛ-21 и ИЛ-2; 3) стадия стабилизации клеточного фенотипа Th17-клеток.
Недавно опубликована очень важная работа, в которой показано, что в механизме образования ИЛ-17 в организме человека существенную роль играют моноциты, которые стимулируют Th17-клетки к продукции ИЛ-17 посредством секреции провоспалительных цитокинов ИЛ-1β и ИЛ-6 [10].
Имеются также сообщения о влиянии микробиоты кишечника на сложные механизмы участия ИЛ-17 в развитии и дальнейшем прогрессировании аутоиммунных заболеваний [4]. Так, установлено, что в патогенезе СД 1-го типа большое значение имеет состояние микрофлоры кишечника, как влияющее на слизистую, содержащую L- и К-клетки, которые секретируют глюкагоноподобный пептид-1 (ГПП-1), так и воздействующее на иммунный статус организма в целом [9].
На основании имеющихся данных высказывается гипотеза, что ключевую роль при воспалительных и аутоиммунных заболеваниях человека (астма, ревматоидный артрит, волчанка, воспалительные заболевания кишечника, множественный склероз, СД и, возможно, злокачественные опухоли) наряду с Th-клетками играет и другая субпопуляция хелперов — регуляторные Th-хелперы (CD4+CD25+FoxP3+-клетки), обладающие противовоспалительным действием. Имеющиеся данные позволяют думать, что в организме здорового человека существует определенный баланс между концентрацией Th17-клеток, секретирующих ИЛ-17, и содержанием регуляторных клеток. При нарушении иммунологической толерантности развивается дисбаланс этих субпопуляций Т-хелперов. Обнаружено, что при аутоиммунных заболеваниях происходит экспансия Th17-клеток, которая сопровождается уменьшением числа и функции Трег (CD4+CD25+FoxP3+) клеток. Сверхэкспрессия FoxP3 приводит к сильной редукции гена ИЛ-17, тормозя RORγt, связанную транскрипцию, путем прямого взаимодействия FoxP3 с RORγt [4, 6, 11].
ИЛ-17 и сахарный диабет 1-го типа
В опытах, проведенных на лабораторных животных со спонтанным аутоиммунным СД (преимущественно на NOD-мышах), неоспоримо доказана ключевая роль ИЛ-17 в патогенезе СД 1-го типа, о чем свидетельствуют данные последних обзоров [4, 6]. Вместе с тем анализ существующих публикаций показывает не всегда полную однозначность результатов, полученных как в эксперименте, так и в клинике, особенно в сложных многоэтапных механизмах развития СД 1-го типа.
Согласно данным N. Martin Orozco et al. (2009), у молодых NOD-мышей в предиабетическом состоянии без ожирения отмечается увеличение экспрессии ИЛ-17А и ИЛ-17F в островках Лангерганса вследствие стремительного развития инсулитов. Блокада же ИЛ-17 уменьшает выраженность воспаления, а соответственно, снижает риск возникновения инсулитов [12]. По результатам же исследований других авторов [13] было обнаружено, что применение анти-ИЛ-17 у молодых (5-недельных) NOD-мышей не оказывает существенного воздействия на возникновение у них СД 1-го типа, в то время как такая же блокада ИЛ-17 у взрослых (10-недельных) NOD-мышей предупреждает развитие диабета (p < 0,01). На основании этого делается заключение, что Th17-клетки играют ведущую роль в развитии СД 1-го типа у NOD-мышей.
При исследовании влияния дефицита одного рецептора ИЛ-17 по сравнению с дефицитом двойного рецептора ИЛ-17/ИФН-γ было установлено, что при дефиците только ИЛ-17 у NOD-мышей наблюдаются более медленное возникновение инсулитов и прогрессирование диабета, в то время как при дефиците ИЛ-17Р/ИФН-γ происходит менее быстрое развитие диабета без появления инсулитов. Причем у животных с двойным дефицитом рецепторов отмечается выраженная лимфоцитопения за счет снижения количества CD4+ Т-клеток. По мнению авторов, полученные результаты подтверждают существующее представление о том, что ИЛ-17/Th17 участвуют в механизме возникновения аутоиммунного диабета, и одновременно показывают, что ИЛ-17 и ИФН-γ могут синергично влиять на механизмы его развития [14].
Важным подтверждением значения Th17-клеток в развитии аутоиммунного диабета у животных явились также исследования, проведенные на другой модели СД 1-го типа — трансгенных мышах NOD/SCID. Было показано, что Th17-клетки, хорошо очищенные от специфических островковых антигенов, способны детерминировать развитие диабета у сингенных иммунодефицитных реципиентов (NOD-SCID-мыши) [15]. В то же время было обнаружено, что при пересадке островков Лангерганса от трансгенных NOD-мышей (BDC/N) мышам-реципиентам (BDC/NScid) у последних происходит быстрое возникновение инсулитов, сопровождаемое повышением уровней транскрипции и количества ИЛ-17 (до 60–80 пг/мл) в сыворотке ПК, которое предшествует развитию диабета [16].
Следует также привести еще одно доказательство участия ИЛ-17 в патогенезе аутоиммунного диабета у животных, в частности, исследования, проведенные на мышах со стрептозотоциновым диабетом, подтверждающие причастность ИЛ-17 к иммунному механизму, приводящему к деструкции бета-клеток [17]. Значительное повышение уровня ИЛ-17 в печени и почках недавно было также обнаружено у крыс с экспериментальным СД по сравнению со здоровыми крысами [18].
Информация о патогенетической роли ИЛ-17 у человека, страдающего СД 1-го типа, немногочисленна и фрагментарна [19]. Во многом она подтверждает данные о ключевой роли этого цитокина в патогенезе СД 1-го типа, полученные в эксперименте на животных. Однако имеются определенные различия патогенеза аутоиммунного СД у экспериментальных животных и человека, особенно касающиеся механизма участия ИЛ-17/Th17-клеток в развитии диабета, что, по-видимому, обусловлено видовой особенностью иммунной системы человека.
Вызывает крайнее удивление тот факт, что публикации о содержании ИЛ-17 в ПК человека на сегодняшний день единичны. По имеющимся данным [20], медиана концентрации ИЛ-17 в ПК 24 пациентов с СД 1-го типа в возрасте от 6 до 30 лет составляла 4,93 (7,37) пг/мл, в то время как у 30 здоровых лиц — 2,61 (7,87) пг/мл. Достоверное увеличение количества секретирующих Th17-клеток было обнаружено у длительно болеющих СД 1-го типа по сравнению со здоровыми лицами контро–льной группы. По данным E.M. Bradshaw и соавт., у больных с впервые выявленным СД 1-го типа отмечалось небольшое, но статистически достоверное повышение Th17-клеток в ПК [21]. Наблюдалось выраженное повышение секреции ИЛ-17 мононуклеарами ПК детей, больных СД 1-го типа [22, 23]. Так, у больных СД 1-го типа медиана содержания ИЛ-17 в мононуклеарах ПК составляла 173 пг/мл, в то время как у здоровых детей ИЛ-17 не определялся вовсе. На основании проведенных исследований авторы приходят к заключению, что повышенная секреция ИЛ-17/ИЛ-22 является главным иммунным нарушением у детей с СД 1-го типа, а следовательно, субпопуляцию Th17-клеток, продуцирующих ИЛ-17, следует считать ведущей в патогенезе СД 1-го типа у человека.
Значительное увеличение числа секретирующих ИЛ-17 CD4+ и CD8+ Т-клеток в ПК детей с впервые выявленным СД 1-го типа было обнаружено A.K. Marwaha и соавт. [10]. Описано также повышение числа циркулирующих CD4+ Т-клеток, продуцирующих ИЛ-17 в ПК, особенно в поджелудочной железе, у пациентов с СД 1-го типа при активации их бета-клеточного антигена моноклональными аутоантителами IA-2A, GADA, включая проинсулин [4, 24].
Как известно [25, 26], у многих больных с длительно протекающим СД 1-го типа в поджелудочной железе остаются функционирующими отдельные бета-клетки. В связи с этим было предпринято сравнительное исследование секретирующих ИЛ-17-клеток у двух групп больных с десятилетним течением СД 1-го типа: тех, у которых сохранялась эндогенная продукция инсулина, и тех, у которых она отсутствовала. Проведенные исследования показали, что у больных с определяемым С-пептидом в отличие от пациентов с неопределяемым С-пептидом имелось достоверное повышение числа ИЛ-17А-клеток (p < 0,001) [27].
Интересно отметить, что повышение экспрессии ИЛ-17А и ИЛ-17F в лимфоцитах ПК наблюдалось и у здоровых людей, которым внутривенно вводили высокие дозы глюкозы, по сравнению со здоровыми лицами контрольной группы, которым глюкозу не вводили [8, 28].
При анализе имеющихся немногочисленных публикаций, касающихся механизма участия ИЛ-17 в возникновении СД 1-го типа у человека, показана его сложность, а самая ранняя стадия еще не полностью установлена. В этом механизме принимают участие многие клеточные элементы естественного и адаптивного иммунитета: различные Т- и В-лимфоциты, нейтрофилы, эозинофилы, моноциты, макрофаги, дендритные и тучные клетки и др. Участие ИЛ-17 и других цитокинов в патогенезе СД 1-го типа схематически сокращенно представлено на рис. 2.
/94-1.jpg)
Еще недавно считалось, что в механизме деструктивного действия островков Лангерганса при СД 1-го типа главную роль играют эффекторные провоспалительные цитокины (ИЛ-1β, ИЛ-6 и ФНО-α), которые продуцируются клоном Th1-клеток, в свою очередь, происходящих из наивных CD4+ Т-клеток путем дифференцировки и воздействия фактора транскрипции STAT4. Th1-клетки под воздействием ИЛ-2 и ИФН-γ затем превращаются в CD8+ Т-клетки, которые обладают цитотоксичной способностью разрушать островковые клетки путем секреции ферментов гранзима и перфорина, расширяющих поры плазматической мембраны клеток [29, 30].
Недавнее открытие нового субкласса CD4+ Т-клеток, т.е. Th17-клеток, секретирующих цитокин ИЛ-17, показало одновременное существование в организме и другого иммунного механизма, способствующего деструкции панкреатических островковых клеток. Как видно из рис. 2, при дифференциации наивных CD4+ Т-клеток наряду с образованием различных их субклонов (Th1, Th2 и др.) под воздействием транскрипционного фактора STAT3 возникает субкласс Th17-клеток, характеризующихся экс–прессией рецепторов транс–крипционных факторов (RORγt или RORC). Под воздействием ТФР-β, ИЛ-6, ИЛ-21, ИЛ-23 происходят дальнейшая пролиферация и дифференциация Th17-клеток и индукция –секреции ИЛ-17, ИЛ-22, ИЛ-23 и CCR6 [4–6].
Полагают, что цитокин ИЛ-17 является одним из главных триггеров, способствующих усилению воспалительной реакции панкреатических островков Лангерганса, благодаря индукции эффекторных провоспалительных цитокинов (ИЛ-1β, ИЛ-6, ФНОα), а также хемокина CCL2.
Важно отметить, что в механизме повышения аутоиммунной деструкции бета-клеток существенную роль играет также действие провоспалительных цитокинов ИЛ-1β, ИЛ-6 и ФНО-α, секретируемых моноцитами, обладающими повышенной функциональной активностью при СД 1-го типа [10]. Эти данные хорошо согласуются с результатами наших исследований [9], в которых показано, что у ОАА-позитивных пациентов в доклиническую и раннюю клиническую стадию развития СД 1-го типа отмечается повышение абсолютного содержания моноцитов в ПК, которые при электронно-микроскопическом и ультрацитохимическом исследовании содержат огромное количество пероксидазоактивных гранул, что указывает на их гиперсекреторную активность.
Значительное место в этом процессе занимает и лептин.
Следовательно, согласно современным представлениям, в патогенезе СД 1-го типа наряду с действием CD8+ Т-клеток, принадлежащих к субклассу Th1 CD4+ Т-клеток, продуцирующих ферменты гранзим и перфорин, которые способствуют деструкции бета-клеток, не менее важную роль играют и Th17-клетки, секретирующие ИЛ-17, которые сингенно с провоспалительными цитокинами (ИЛ-1β, ИЛ-6 и ФНО-α) усиливают воспаление островков Лангерганса. При этом происходит резкое повышение окиси азота (увеличение экспрессии супероксида дисмутазы-2, синтазы-2А, окиси азота и циклооксигеназы-2), выделение свободных радикалов, апоптоз и деструкция бета-клеток, что завершается клинической манифестацией диабета [22, 23].
Вместе с тем в организме животных и человека существует и протекторный иммунный механизм, препятствующий аутоиммунной деструкции панкреатических бета-клеток и развитию СД 1-го типа, который осуществляется путем супрессии Th17-клеток. Главенствующая роль в этом механизме принадлежит особому субклассу CD4+ Т-клеток, получивших название Т-регуляторных (Tрег). Этот субкласс CD4+ Т-клеток идентифицируется как CD4+CD25+Foxp3+-лимфоциты, экспрессирующие на поверхности клеток CD25-антиген и транскрипционный регулятор Foxp3 (рис. 1, 2). Трег-клетки также происходят из наивных CD4+ Т-клеток путем их дифференцировки с помощью транскрипционных факторов STAT5 и FoxР3 и соответствующих цитокиновых сигналов [5, 31].
CD4+CD25+Foxp3-клетки составляют 5–10 % CD4+-клеток у человека и являются неотъемлемой частью системы периферической толерантности и константного гомеостаза. Большинство Tрег-клеток находится в тимусе (тТрег) и, перемещаясь на периферию, становится пTрег. Некоторые Tрег-клетки развиваются из конвенциональных CD4+-клеток при реакции на антиген и идентифицируются как индуцированные или антигенспецифичные (иTрег). Tрег-клетки играют ключевую роль в иммунообусловленной аутотолерантности, что обеспечивает защитное действие против различных патогенов, обладает онкопротекторной способностью, а также тормозит отторжение трансплантата. Для выживания и пролиферации Tрег-клеткам необходима достаточная секреция ИЛ-2, а также одновременное подавление функционирования CD4+CD25–-клеток. Этот субкласс CD4+ Т-клеток может кооперироваться с другими гетерогенными клонами Th1-, Th2-, Th17-клеток. Одновременно показано, что функция Tрег-клеток частично или полностью определяется также различными видами цитокиновых сигналов. Tрег-клетки обладают большой фенотипической пластичностью и мимикрирующей поляризацией, которые во многом зависят от микроокружения. При потере аллотолерантности может происходить патологическая конверсия Tрег-клеток в exFoxp3. Tрег-клетки при этом становятся нестабильными и дисфункциональными в присутствии провоспалительных цитокинов. Кроме того, эти клетки экспрессируют рецептор транс–крипционного фактора RORγt и воспалительные цитокины, которые тормозят их иммуносупрессивные свойства, индуцируя продукцию цитокинов ИЛ-17. Имеются неоспоримые доказательства того, что Tрег-клетки тормозят воспалительный процесс в островках Лангерганса, апоптоз и деструкцию бета-клеток, которые сопровождаются уменьшением стабильности экспрессии FoxР3 и увеличением пропорции Th1-подобных клеток в финале развития СД1Т. Недавно было также показано, что в механизме этого процесса существенную роль играет c-Jun N-terminal kinase-1, которая тормозит иммунные воспалительные процессы. Параллельно при этом также происходит повышение продукции протекторных цитокинов ИЛ-4 и ИЛ-10 [31, 32].
Интересно отметить, что при инкубации in vitro высокоочищенных CD4+-клеток только с ТФР-β появляются Трег-клетки, в то время как при прибавлении в культуру ТФР-β+ ИЛ-6 происходит образование Th17-клона [33]. Естественно, в организме in vivo дифференциация и пролиферация ИЛ-17 из наивных CD+ Т-клеток происходят более сложным путем с участием, как уже указывалось, многих видов цитокинов и хемокинов.
Согласно имеющимся данным, на сегодняшний день ряд авторов [6, 11] выдвигают гипотезу о том, что у здорового человека существует позитивный баланс между содержанием Th17-клеток и CD4+CD25+Foxp3+T-клетками (Трег-клетками). Развитие СД 1-го типа является результатом возникновения дисбаланса между этими двумя видами CD4+-клеток, т.е. происходит в результате превалирования Th17-клеток с гиперсекрецией ИЛ-17, особенно сингенно с клоном Th1-клеток, секретирующим провоспалительные цитокины.
ИЛ-17 и сахарный диабет 2-го типа
При анализе исследований, проведенных на лабораторных животных и больных СД 2-го типа, у большинства из них была обнаружена позитивная корреляция между количеством Th17-клеток, уровнем ИЛ-17 в ПК, с одной стороны, и развитием инсулиновой резистентности (ИР), возникновением СД 2-го типа и его осложнений — с другой [1, 6, 34]. Так, C. Chen и соавт. из известной клиники Mayo (США) обнаружили значительное повышение уровня ИЛ-17 в сыворотке ПК у больных с впервые выявленным СД 2-го типа по сравнению с группой здоровых лиц (10,44 ± 6,47 пг/мл против 2,99 ± 1,68 пг/мл, p < 0,01), которое сопровождалось повышением транскрипционного фактора RORγt в мононуклеарах ПК (p < 0,01) [35]. Достоверное повышение уровня ИЛ-17 одновременно с увеличением содержания ИЛ-22 и провоспалительных цитокинов ИЛ-β и ИЛ-6 в сыворотке ПК больных СД 2-го типа наблюдали N. Fatima и соавт. [34]. Причем наиболее выраженное изменение отмечалось у больных СД 2-го типа старше 51 года. Аналогичное повышение уровня ИЛ-17 в сыворотке ПК больных СД 2-го типа описано и другими авторами [36, 37].
В опытах на мышиной модели СД 2-го типа было показано, что при применении ИЛ-17-антител, при котором полностью нейтрализуется уровень ИЛ-17 в циркуляции, одновременно происходят снижение уровня сывороточного ФНО-α и повышение антивоспалительного цитокина адипонектина. В результате этого заметно тормозится развитие ИР и СД 2-го типа, при этом происходит ослабление генов провоспалительных цитокиновых сигналов, регулирующих уровень инсулина в организме, что способствует развитию ИР, а затем и СД 2-го типа [38].
Повышение продукции и уровня ИЛ-17 в циркуляции описано также и при характерных осложнениях у больных СД 2-го типа: кардиоваскулярных, периодонтальных, ожирении и т.д. [7, 36, 39, 40].
Особенно большой интерес представляет выяс–нение причин повышения уровня ИЛ-17 у больных СД 2-го типа, сопровождаемого ожирением или избыточной массой тела, так как ожирение присутствует у 80 % больных СД 2-го типа [41, 42] и является неотъемлемой составляющей метаболического синдрома [43]. В настоящее время считается абсолютно доказанной ведущая роль ИЛ-17 в процессах, сопровождающих развитие ожирения. При ожирении, считающемся одним из видов воспаления жировой ткани, ее дендритные клетки активируют лептин и МИФ (фактор, тормозящий миграцию макрофагов), в результате чего происходит стимуляция продукции ИЛ-17, воздействующая на ядерный фактор NF-κB, который индуцирует экспрессию генов провоспалительных цитокинов [44].
В связи с этим возникает вопрос: является ли это повышение секреции ИЛ-17 следствием аутоиммунного процесса и низкоинтенсивного воспаления, лежащих в основе развития СД 2-го типа [44, 45], или самостоятельным независимым явлением, или же имеющим сингенно-сочетанный характер (СД 2-го типа + ожирение)? Естественно, для ответа на этот вопрос необходимы дальнейшие исследования в будущем.
Приведенные выше исследования являются убедительным доказательством того, что ИЛ-17 играет ключевую роль в развитии ИР и СД 2-го типа у человека. Все же механизм участия ИЛ-17 в возникновении СД 2-го типа, как и СД 1-го типа, во многом еще не совсем ясен. Полагают, что в механизме неблагоприятного действия ИЛ-17 при СД 2-го типа большую роль играет активация ИЛ-17 ядерного фактора каппа (NF-κB), стимулирующего экспрессию генов воспалительных цитокинов (ИЛ-1β, ИЛ-6, ФНО-α), которые индуцируют ИР и ведут к развитию клинического дебюта СД [6].
В настоящее время мы находимся только в начале пути изучения биологической и патогенетической роли ИЛ-17 в организме человека. Все же, согласно уже имеющимся на сегодняшний день данным, бесспорно, что ИЛ-17 оказывает мощное потенцирующее влияние на течение воспалительного процесса в островках Лангерганса путем стимулирующей продукции провоспалительных цитокинов и хемокинов, а также ускорения деструкции бета-клеток в результате патологических нарушений естественного и адаптивного иммунитета, приводящих к развитию СД 1-го типа. Более полная информация об ИЛ-17 в будущем поможет в создании новых научно обоснованных путей для разработки методов пред–отвращения, терапии и профилактики СД.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages [Text] / L.E. Harrington, R.D. Hatton, P.R. Mangan [et al.] // Nat. Immunol. — 2005. — Vol. 6, № 11. — P. 1123-1132.
2. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17 [Text] / H. Park, Z. Li, X.O. Yang [et al.] // Nat. Immunol. — 2005. — Vol. 6, № 11. — P. 1133-1141.
3. IL-22 and IL-17: An overview [Text] / R. Sabat, E. Witte, K. Witte, K. Wolk // Chapter from book IL-17, IL-22 and their producing cells: Role in inflammation and autoimmunity. — SpringerLink, 2013. — Р. 11-35.
4. Li Y. Th17 Cells in type 1 diabetes: role in the pathogenesis and regulation by gut microbiome [Text] / Y. Li, Y. Liu, C.Q. Chu // Mediators Inflamm. — 2015. — 2015. — 638470.
5. O’Shea J.J. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells [Text] / J.J. O’Shea, W.E. Paul // Science. — 2010. — Vol. 327, № 5969. — P. 1098-1102.
6. Abdel-Moneim A. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus [Text] / A. Abdel-Moneim, H.H. Bakery, G. Allam // Biomed. Pharmacother. — 2018. — Vol. 101. — P. 287-292.
7. Chehimi M. Pathogenic role of IL-17-producing immune cells in obesity, and related inflammatory diseases [Text] / M. Chehimi, H. Vidal, A. Eljaafari // J. Clin. Med. — 2017. — Vol. 6, № 7. — P. pii:E68.
8. Kumar P. Molecular underpinnings of Th17 immune-regulation and their implications in autoimmune diabetes [Text] / P. Kumar, G. Subramaniyam // Cytokine. — 2015. — Vol. 71, № 2. — P. 366-376.
9. Сахарный диабет. Иммунитет. Цитокины [Текст] / К.П. Зак, Н.Д. Тронько, В.В. Попова, А.К. Бутенко. — К.: Книга плюс, 2015. — 485 с.
10. Cutting edge: Increased IL-17-secreting T cells in children with new-onset type 1 diabetes [Text] / A.K. Marwaha, S.Q. Crome, C. Panagiotopoulos [et al.] // J. Immunol. — 2010. — Vol. 185, № 7. — P. 3814-3818.
11. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes / A. Ferraro, C. Socci, A. Stabilini [et al.] [Text] // Diabetes. — 2011. — Vol. 60, № 11. — P. 2903-2913.
12. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells [Text] / N. Martin-Orozco, Y. Chung, S.H. Chang [et al.] // Eur. J. Immunol. — 2009. — Vol. 39, № 1. — P. 216-224.
13. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice [Text] / J.A. Emamaullee, J. Davis, S. Merani [et al.] // Diabetes. — 2009. — Vol. 58, № 6. — P. 1302-1311.
14. Double deficiency in IL-17 and IFN-γ signalling significantly suppresses the development of diabetes in the NOD mouse [Text] / G. Kuriya, T. Uchida, S. Akazawa [et al.] // Diabetologia. — 2013. — Vol. 56, № 8. — P. 1773-1780.
15. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice [Text] / D. Bending, H. De la Peña, M. Veldhoen [et al.] // J. Clin. Invest. — 2009. — Vol. 119, № 3. — P. 565-572.
16. Dynamic interaction between T cell-mediated beta-cell da–mage and beta-cell repair in the run up to autoimmune diabetes of the NOD mouse [Text] / S.S. Vukkadapu, J.M. Belli, K. Ishii [et al.] // Physiol. Genomics. — 2005. — Vol. 21, № 2. — P. 201-211.
17. Interleukin-17A deficiency ameliorates streptozotocin-induced diabetes [Text] / Z. Tong, W. Liu, H. Yan, Ch. Dong // Immunology. — 2015. — Vol. 146, № 2. — P. 339-346.
18. Diabetes increases interleukin-17 levels in periapical, hepatic, and renal tissues in rats [Text] / M.M. Azuma, J.E. Gomes-Filho, A.K.C. Prieto [et al.] // Arch. Oral. Biol. — 2017. — Vol. 83. — P. 230-235.
19. Th17 pathway in recent-onset autoimmune diabetes [Text] / J.P. Fores, L.G. Crisostomo, N.M. Orii [et al.] // Cell. Immunol. — 2018. — Vol. 324. — P. 8-13.
20. Serum IL-17, IL-23, and TGF-β levels in type 1 and type 2 diabetic patients and age-matched healthy controls [Text] / A. Roohi, M. Tabrizi, F. Abbasi [et al.] // Biomed. Res. Int. — 2014. — 2014. — 718946.
21. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells [Text] / E.M. Bradshaw, K. Raddassi, W. Elyaman [et al.] // J. Immunol. — 2009. — Vol. 83, № 7. — P. 4432-4439.
22. IL-17 immunity in human type 1 diabetes [Text] / J. Honkanen, J.K. Nieminen, R. Gao [et al.] // J. Immunol. — 2010. — Vol. 185, № 3. — P. 1959-1967.
23. Coxsackievirus up-regulates IL-17 immunity in human type 1 diabetes [Text] / J.K. Honkanen, J.K. Nieminen, S. Skarsvik [et al.] // Diabetologia. — 2011. — Vol. 54, Suppl. 1 (B). — A-421.
24. Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated в-cell death [Text] / S. Arif F., K. Marks, T. Bouckenooghe [et al.] // Diabetes. — 2011. — Vol. 60, № 8. — P. 2112-2119.
25. Зак К.П. Иммунные и противовоспалительные факторы в механизме лечебного действия метформина [Текст] / К.П. Зак, О.В. Фурманова // Міжнародний ендокринологічний журнал. — 2018. — Т. 14, № 2. — С. 90-97.
26. Current concepts on the pathogenesis of type 1 diabetes — considerations for attempts to prevent and reverse the disease [Text] / M.A. Atkinson, M. von Herrath, A.C. Powers, M. Clare-Salzler // Diabetes Care. — 2015. — Vol. 38, № 6. — P. 979-988.
27. Increased interleukin-35 levels in patients with type 1 diabetes with remaining C-peptide [Text] / D. Espes, K. Singh, S. Sandler, P.O. Carlsson // Diabetes Care. — 2017. — Vol. 40, № 8. — P. 1090-1095.
28. Kumar P. High glucose driven expression of pro-inflammatory cytokine and chemokine genes in lymphocytes: molecular mechanisms of IL-17 family gene expression [Text] / P. Kumar, K. Natarajan, N. Shanmugam // Cell Signal. — 2014. — Vol. 26, № 3. — P. 528-539.
29. Mandrup-Poulsen T. Interleukin-1 antagonism: a study companion for immune tolerance induction in type 1 diabetes? [Text] / T. Mandrup-Poulsen // Diabetes. — 2014. — Vol. 63, № 6. — P. 1833-1835.
30. Rabinovich A. Roles of cytokines in the pathogenesis and therapy of type 1 diabetes [Text] / A. Rabinovich, W.L. Suarez-Pinzon // Cell. Biochem. Biophys. — 2007. — Vol. 48, № 2–3. — P. 159-163.
31. С-Jun N-terminal kinase 1 defective CD4+CD25+ FoxP3+ cells prolong islet allograft survival in diabetic mice [Text] / D. Tripathi, S.S. Cheekatla, P. Paidipally [et al.] // Sci. Rep. — 2018. — Vol. 19, № 8(1). — P. 3310.
32. Pathological conversion of regulatory T cells is associated with loss of allotolerance [Text] / J. Hua, T. Inomata, Y. Chen [et al.] // Sci. Rep. — 2018. — Vol. 8, № 1. — P. 7059.
33. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells [Text] / E. Bettelli, Y. Carrier, W. Gao [et al.] // Nature. — 2006. — Vol. 441, № 7090. — P. 235-238.
34. Emerging role of Interleukins IL-23/IL-17 axis and biochemical markers in the pathogenesis of type 2 diabetes: Association with age and gender in human subjects [Text] / N. Fatima, S.M. Faisal, S. Zubair [et al.] // Int. J. Biol. Macromol. — 2017. — Vol. 105(Pt. 1). — P. 1279-1288.
35. Elevated interleukin-17 levels in patients with newly diag–nosed type 2 diabetes mellitus [Text] / C. Chen, Y. Shao, X. Wu [et al.] // Biochem. Physiol. — 2016. — Vol. 5, № 206. — P. 2-10.
36. Effect of nonsurgical periodontal therapy on plasma le–vels of IL-17 in chronic periodontitis patients with well controlled type-II diabetes mellitus — a clinical study [Text] / J.V. Sunandhakumari, A. Sadasivan, E. Koshi [et al.] // Dent. J. (Basel). — 2018. — Vol. 6, № 2. — P. pii: E19.
37. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy [Text] / C. Zhang, C. Xiao, P. Wang [et al.] // Hum. Immunol. — 2014. — Vol. 75, № 4. — P. 289-296.
38. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance [Text] / K. Ohshima, M. Mogi, F. Jing [et al.] // Hypertension. — 2012. — Vol. 59, № 2. — P. 493-499.
39. Interleukin-17A gene variability in patients with type 1 diabetes mellitus and chronic periodontitis: Its correlation with IL-17 levels and the occurrence of Periodontopathic Bacteria [Text] / P. Borilova Linhartova, J. Kastovsky, S. Lucanova [et al.] // Mediators Inflamm. — 2016. — 2016. — 2979846.
40. The ratios of pro-inflammatory to anti-inflammatory cytokines in the serum of chronic periodontitis patients with and wi–thout type 2 diabetes and/or smoking habit [Text] / T.S. Miranda, S.L. Heluy, D.F. Cruz [et al.] // Clin. Oral. Invest. — 2018 May.
41. Тронько Н.Д. Ожирение и сахарный диабет / Н.Д. Тронько, К.П. Зак // Лікарська справа. — 2013. — Т. 8, № 1125. — С. 3-21.
42. Obesity is associated with poorer clinical outcomes following insulin initiation for patients with type 2 diabetes [Text] / S. Kumar, B. Wilson, L. Watson, J. Alsop // Diabetologia. — 2009. — Vol. 52 (suppl. 1).
43. Зак К.П. Иммунитет у больных сахарным диабетом 2 и сопутствующим метаболическим синдромом. Сообщение 2. Роль адипонектинов (ИЛ-6, ФНОα, лептина и адипонектина) / К.П. Зак, Б.Н. Маньковский, И.Н. Кондрацкая // Ендокринологія. — 2013. — Т. 18, № 2. — С. 26-32.
44. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? [Text] / S. Tsai, X. Clemente-Casares, X.S. Revelo [et al.] // Diabetes. — 2015. — Vol. 64, № 6. — P. 1886-1897.
45. Donath M.Y. What is the role of autoimmunity in type 1 diabetes? A clinical perspective [Text] / M.Y. Donath, C. Hess, E. Palmer // Diabetologia. — 2014. — Vol. 57, № 4. — P. 653-655.