Резюме
У статі подано власне клінічне спостереження синдрому мікроделеції 22q11.2 хромосоми в дитини з природженою вадою серця, фенотиповими ознаками її також були аномалії лицьового черепа, імунні порушення, затримка розвитку та навіть когнітивний дефіцит. Для діагностики даного синдрому авторами проведено аналіз сучасних світових критеріїв визначення, стандартів діагностики та моніторингу хворих. У роботі визначена епідеміологія синдрому делеції 22q11.2, що зустрічається з частотою від 1 : 3000 до 1 : 6000 новонароджених і успадковується автосомно-домінантним способом, проте в Україні синдром діагностується рідко, що, на нашу думку, обумовлено відсутністю неонатального скринінгу й недостатньою обізнаністю лікарів щодо клінічних особливостей хвороби. Ділянка 22q11.2 є однією з найбільш структурно складних у геномі, передусім через декілька великих блоків. Локус LCR (low copy repeats) регіону 22q11.2 вразливий до генетичної помилки, що обумовлено 96% ідентичністю. Акцентовано увагу на тому, що симптоми клінічної маніфестації варіюють залежно від віку. У дітей раннього віку типові симптоми включають комбінацію природженої вади серця, імунодефіциту, мальформації верхнього піднебіння, гіпокальціємію, труднощі в годуванні, затримку психічного й мовленнєвого розвитку, розлади поведінки, аномалії будови нирок та статевих органів, ларинготрахеоезофагальні мальформації, гіпотиреоїдизм, порушення будови скелета. У деяких дітей захворювання маніфестує в шкільному віці аномаліями поведінки, затримкою психічного розвитку й гіпокальціємією. Наявність характерних рис обличчя може сприяти ідентифікації синдрому в будь-якому віці. Водночас труднощі діагностики пов’язані з широкою фенотиповою мінливістю. У статті детально описана генетична діагностика синдрому за декількома методиками (флуоресцентна гібридизація in situ, мультиплексна лігазна ланцюгова реакція, застосування хромосомного мікрочіпу), вибір яких залежить від періоду життя та вираженості фенотипових ознак. Для моніторингу стану дитини із синдромом мікроделеції 22q11.2 хромосоми застосовуються методики «багатопрофільний командний підхід» і «система за системою».
В статье представлено собственное клиническое наблюдение синдрома микроделеции 22q11.2 хромосомы у ребенка с врожденным пороком сердца, фенотипическими признаками которого были также аномалии лицевого черепа, иммунные нарушения, задержка развития и даже когнитивный дефицит. Для диагностики данного синдрома авторами проведен анализ современных мировых критериев определения, стандартов диагностики и мониторинга больных. В работе определена эпидемиология синдрома делеции 22q11.2, который встречается с частотой от 1 : 3000 до 1 : 6000 новорожденных и наследуется аутосомно-доминантным способом, однако в Украине синдром диагностируется редко, что, по нашему мнению, обусловлено отсутствием неонатального скрининга и недостаточной осведомленностью врачей относительно клинических особенностей болезни. Участок 22q11.2 является одним из наиболее структурно сложных участков генома, прежде всего из-за нескольких больших блоков. Локус LCR (low copy repeats) региона 22q11.2 уязвим к генетической ошибке, что обусловлено 96% идентичностью. Акцентировано внимание на том, что симптомы клинической манифестации варьируют в зависимости от возраста. У детей раннего возраста типичные симптомы включают комбинацию врожденного порока сердца, иммунодефицита, мальформации верхнего неба, гипокальциемии, трудности в кормлении, задержку психического и речевого развития, расстройства поведения, аномалии строения почек и половых органов, ларинготрахеоэзофагеальные мальформации, гипотиреоидизм, нарушение строения скелета. У некоторых детей заболевание манифестирует в школьном возрасте аномалиями поведения, задержкой психического развития и гипокальциемией. Наличие характерных черт лица может способствовать идентификации синдрома в любом возрасте. В то же время трудности в диагностике связаны с широкой фенотипической изменчивостью. В статье подробно описана генетическая диагностика синдрома по нескольким методикам (флуоресцентная гибридизация in situ, мультиплексная лигазная цепная реакция, применение хромосомного микрочипа), выбор которых зависит от периода жизни и выраженности фенотипических признаков. Для мониторинга состояния ребенка с синдромом микроделеции 22q11.2 хромосомы используются методики «многопрофильный командный подход» и «система за системой».
The article presents own clinical observation of the chromosome 22q11.2 microdeletion syndrome in a child with congenital heart disease. Phenotype also includes facial skull anomalies, immune disorders, developmental delay and even cognitive deficits. The authors analyzed the current world definition criteria, standards for diagnosis and monitoring of patients to diagnose this syndrome. The epidemiology of 22q11.2 deletion syndrome is determined in the work. Its incidence varies between 1 : 3,000–1 : 6,000 newborns, the inheritance is autosomal dominant, however, in Ukraine the syndrome is rarely diagnosed, which in our opinion is due to the lack of neonatal screening and insufficient awareness of physicians of clinical features of the disease. Section 22q11.2 is one of the most structurally complex regions of the genome, primarily through several large blocks. The locus of low copy repeats in region 22q11.2 is prone to a genetic error, which is due to 96% identity. Attention is focused that clinical manifestations vary with age. In young children, typical symptoms include a combination of congenital heart disease, immunodeficiency, malformation of the palate, hypocalcaemia, difficulty in feeding, delayed mental and speech development, behavioral disorders, kidney and genital abnormalities, laryngo-tracheo-esophageal malformations, hypothyroidism, skeletal dysmorphology. In some children, the disease manifests in school-age by behavioral anomalies and mental retardation, and hypocalcaemia. The presence of facial features can contribute to identifying the syndrome at any age. At the same time, difficulties in diagnosis are associated with widespread phenotypic variability. The article describes in detail the genetic diagnosis of the syndrome using several methods (fluorescence hybridization in situ, multiplex ligation-dependent probe amplification and chromosomal microarray), the choice of which depends on the period of life and the expressiveness of phenotypic traits. To monitor the child with the syndrome of chromosome 22q11.2 microdeletion, the authors describe approaches that include multidisciplinary team approach and system after system method.
Синдром делеції 22q11.2 хромосоми є найбільш поширеним синдромом мікроделеції з поліорганною дисфункцією. Фенотиповими ознаками синдрому вважаються вади серця та судин, аномалії лицьового черепа, імунні, ендокринні, сечостатеві та шлунково-кишкові порушення, затримка розвитку, когнітивний дефіцит та навіть нейропсихічні захворювання (наприклад, шизофренія) [1, 2]. Епонімічний термін «синдром Ді Джорджі» був визначений в 1965 році, синдром включав аплазію тимуса й паращитоподібних залоз. Природжені вади серця та судин в структуру синдрому увійшли пізніше у зв’язку з визнанням теорії порушення розвитку третьої та четвертої зябрових дуг протягом ембріогенезу. Доведена асоціація фенотипу із цукровим діабетом у матері, а також мутацією в модифікаторі організації хроматину хелікази ДНК-зв’язуючого протеїну 7 (CHD), що відома як CHARGE-синдром, або в T-box 1 (TBX1) гені. У 1980-х рр. доведено, що делеція 22q11.2 є етіологічним фактором синдрому Ді Джорджі. На початку 90-х років упроваджена методика флуоресцентної гібридизації in situ (fluorescence in situ hybridization — FISH) з використанням зондів у межах ділянки 22q11.2, що деталізувала субмікроскопічні делеції 22q11.2 як чинники синдрому Ді Джорджі [1, 3].
Власне клінічне спостереження
Хлопчик Л., 4 роки (рис. 1), перебуває на диспансерному спостереженні в Обласній клінічній дитячій лікарні.
З анамнезу життя: дитина від І вагітності, що перебігала на тлі еклампсії в матері. Множинні вади розвитку не діагностовано пренатально. Пологи шляхом кесарева розтину в терміні гестації 36 тижнів, маса тіла 2600 г. Природжена вада серця (дефект міжшлуночкової перегородки, відкрита артеріальна протока) та вроджені вади кістково-м’язової системи (природжена розщелина верхньої губи зліва, повне розщеплення твердого й м’якого піднебіння, викривлення носової перегородки) діагностовано в ранньому неонатальному періоді.
При ультразвуковій діагностиці тимус не візуалізувався. Імунограма у віці 6 місяців: абсолютна кількість лімфоцитів — 2,02 (5,4–7,59) • 109/л, CD3 — 1,33 (1,7–3,6) • 109/л, CD4 — 33 % (38–50 %), CD19 — 0,42 (0,5–1,5) • 109/л, CD16 — 20 % (8–17 %), IgA — 1,1 (0,37 ± 0,18) г/л, IgA — 1,1 (0,37 ± 0,18) г/л, IgМ — 0,9 (0,54 ± 0,23) г/л, IgG — 10,0 (6,61 ± 0,69) г/л.
Рівень іонізованого кальцію сироватки крові 1,32 ммоль/л (норма).
Діагностика синдрому делеції 22q11.2 методом FISH: виявлена типова делеція 22q11.2 — LCR22A.
У віці 2 місяці проведена радикальна операція з приводу природженої вади серця. Післяопераційний період ускладнився пневмонією, яка супроводжувалась торпідним перебігом та стійким синдромом бронхіальної обструкції, що дало підставу для проведення поглибленого діагностичного пошуку аномалії будови трахеобронхіального дерева й первинного імунодефіциту.
Припущення щодо бронхомаляції підтверджено при бронхоскопічному дослідженні. Окрім виявлених мальформацій, у дитини спостерігалися асиметрія обличчя під час посмішки або плачу, аурикулярні аномалії та характерні риси обличчя, такі як нависання повік і широке перенісся. При ультразвуковій діагностиці тимус не візуалізувався, за даними імунограми виявлений Т-клітинний імунодефіцит. Отримані дані дали підставу припустити наявність синдрому делеції 22q11.2, що було підтверджено методом FISH: виявлена типова делеція 22q11.2 — LCR22A.
Згодом дитині проведені поетапні радикальні операції з приводу краніофаціальних аномалій в один та два роки. Для моніторингу стану дитини залучені методики «система за системою» та «багатопрофільний командний підхід». При черговому огляді в 4 роки: дитина затримується в психічному розвитку, водночас має прогрес в елементарному навчанні (рис. 1). Хлопчик здатний вимовляти лише деякі звуки, проте з успіхом проходить спеціалізоване навчання в логопеда. Вакцинація проводиться за індивідуально розробленим графіком, без використання живих вакцин; 1 раз на 6 місяців проводиться оцінка імунної реактивності з її корекцією. Сім’я хлопчика обізнана щодо необхідності консультації генетика при плануванні наступних вагітностей.
Отже, клінічне спостереження демонструє класичний фенотип синдрому делеції 22q11.2, що включає краніофасціальні аномалії, природжену ваду серця, аплазію тимуса та характерні риси обличчя. Водночас унікальність випадку демонструє наявність бронхомаляції, що спостерігається рідко й може поповнити базу фенотипових ознак синдрому делеції 22q11.2.
Обговорення
Сьогодні різноманітні фенотипи делеції 22q11.2 хромосоми асоціюються із шістьма синдромами:
1) синдром Ді Джорджі;
2) велокардіофасціальний синдром;
3) синдром конотрункальних вад та аномалій обличчя;
4) автосомно-домінантний Opitz G/BBB синдром;
5) синдром Седлакова;
6) кардіофасціальний синдром Кайлера.
Епідеміологія. За даними популяційних досліджень 1990–2000 рр., частота синдрому делеції 22q11.2 коливається в межах 1 : 3000 — 1 : 6000 новонароджених, він успадковується автономно-домінантним способом [3, 4]. Близько 93 % пробандів мають делецію de novo з 22q11.2 і 7 % — успадковану від батьків. Популяції уражених людей мають 50% шанс наслідувати делеції 22q11.2. В Україні синдром діагностується рідко, що, на нашу думку, обумовлено відсутністю неонатального скринінгу й недостатньою обізнаністю лікарів у клінічних особливостях хвороби. Водночас ймовірність синдрому може визначатися за фенотиповими ознаками. Більшість новонароджених з синдромом 22q11.2 делеції мають природжену ваду серця, рідше — мальформації твердого піднебіння, затримку розвитку, згодом шизофренію [3]. Поширеність гіпопаратиреозу не уточнена. Пацієнти із синдромом делеції 22q11.2 мають високий ризик неонатальної смертності, –обумовлений станами, пов’язаними з природженими вадами серця, аспіраційним синдромом (за рахунок мальформації твердого піднебіння), гіпокальціємічними судомами (обумовленими гіпопаратиреозом), трахеобронхомаляцією. Останніми роками значно покращилися можливості хірургічної корекції природжених вад серця, і хворі виживають до фертильного віку й можуть мати дітей з підвищеним ризиком формування природженої вади серця [3].
У 2012–2015 рр. у результаті двох багатоцентрових пренатальних досліджень з використанням методів інвазійного пренатального скринінгу плодів виявлені 22q11.2 делеції у 1 : 347 — 1 : 992 плодів відповідно. Делеція 22q11.2 виявлялася в кожного сотого плода з природженою вада серця й у кожного тисячного — з нормальною будовою тіла за даними ультразвукового дослідження. У кожного 170-го новонародженого діагностовано синдромом делеції 22q11 з ймовірністю відхилення нейропсихічного й фізичного розвитку. Вірогідної залежності частоти захворювання від статі, расової належності, етнічних груп не виявлено [1].
Етіологія. Типові делеції 22q11.2 (LCR22A та LCR22D), що діагностуються методом флуоресцентної гібридизації іn situ, включають 30–40 генів, три мільйони ДНК на одній з пар 22-ї хромосоми в кожній клітині. Нетипові делеції на 22-й хромосомі (LCR22B-LCR22D і LCR22C-LCR22D) зазвичай перебігають у стертій формі та не виявляються флуоресцентною гібридизацією іn situ. Сьогодні більшість пацієнтів (90–95 %) мають делеції 22q11.2 de novo, проте очікується зростання частоти спадкових варіантів з огляду на збільшення тривалості життя людей, які досягають репродуктивного віку [5, 6].
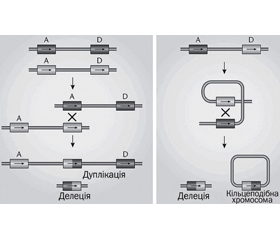
Патофізіологія. Ділянка 22q11.2 є однією з найбільш структурно складних ділянок геному, у першу чергу через декілька великих блоків. Локус LCR (low copy repeats) регіону 22q11.2 вразливий до генетичної помилки, що обумовлено 96% ідентичністю. 85 % пацієнтів мають делеції в LCR22A та LCR22D як результат неалельної гомологічної рекомбінації між LCR22A та LCR22D [1] (рис. 2).
У локусі делеції розташовано 46 генів, що кодують протеїни. TBX1 — найбільш вивчений ген у регіоні делеції 22q11.2, гетерозиготи з мутаціями TBX1 мають кардіоваскулярні дефекти, аплазію або гіпоплазію тимуса та паращитоподібних залоз, гетерозиготи за геном DGCR8 — вади розвитку нервової системи. Існують і інші мутації генів LCRs 22q11.2, пов’язані із захворюваннями, щодо більшості яких тривають дослідження на моделях тварин [6, 7].
Онтогенез людини за умови делеції 22q11.2 (рис. 3). Значна частина мальформацій та порушень функції при делеції 22q11.2 пов’язана з морфогенезом зябрових дуг: черепно-лицьові аномалії, вади серецево-судинної системи, аплазія та гіпоплазія тимуса й паращитоподібних залоз. Ці структури формуються за рахунок усіх трьох класичних зародкових шарів ембріона — ендодерми, мезодерми, ектодерми разом з клітинами нервового гребеня. Краніофасціальні м’язи, серце, аорта та легенева артерія — з глоткової мезодерми; кістки обличчя та твердого піднебіння — з клітин нервового гребеня та передньої мезодерми. Паращитоподібна залоза й тимус виникають унаслідок взаємодії тканин між ендодермою глотки та клітинами нервового гребеня [8, 9].
/158-1.jpg)
Шляхи формування серця, тимуса, паращитоподібних залоз і мозку за участю TBX1 генів подані на рис. 4 [1]. Делеція TBX1 порушує регуляцію сигнальних шляхів, сприяє порушенню міграції кардіальних клітин нервового гребеня (Cardiac neural crest cells). Серце, тимус та паращітоподібні залози закладаються аномально.
/159-1.jpg)
Клінічні особливості. Симптоми клінічної маніфестації варіюють залежно від віку дитини. У дітей раннього віку типові симптоми включають комбінацію природженої вади серця, імунодефіциту, мальформації верхнього піднебіння, гіпокальціємію, труднощі в годуванні, затримку психічного й мовленнєвого розвитку, розлади поведінки, аномалії будови нирок та статевих органів, ларинготрахеоезофагальні мальформації, гіпотиреоїдизм, порушення будови скелета. У шкільному віці захворювання маніфестує аномаліями поведінки, що асоційовано із затримкою психічного розвитку та гіпокальціємією. Наявність характерних рис обличчя може сприяти ідентифікації синдрому в будь-якому віці. Водночас труднощі в діагностиці в підлітків та дорослих людей пов’язані з широкою фенотиповою мінливістю. Часто генетичне дослідження в дорослих проводиться після народження постраждалого дитини з фенотипом делеції 22q11.2. У табл. 1 ми наводимо основні аномалії, характерні для даного синдрому [1, 10–21].
/159-2.jpg)
Серцево-судинні аномалії. Серцево-судинні аномалії часті, вони можуть проявлятися тетрадою Фалло (з атрезією легеневої артерії або без неї) (~20 % пацієнтів), переривом дуги аорти (~13 % пацієнтів), дефектом міжшлуночкової перегородки (~14 % пацієнтів), загальним артеріальним стовбуром (~6 % пацієнтів), судинним кільцем (~5,5 % пацієнтів), дефектом міжпередсердної перегородки (~14 % пацієнтів) та іншими природженими вадами серця (~10 % пацієнтів). Водночас у 24 % дітей із синдромом делеції 22q11.2 мальформації серцево-судинної системи можуть бути відсутні [1, 10–21].
Імунна недостатність. За даними статистики, 77 % пацієнтів із синдромом делеції 22q11.2 віком понад 6 місяців мають дефіцит імунної системи; у 67 % продукування Т-клітин було порушене, у 19 % виявлена комбінація клітинного та гуморального дефектів, у 23 % спостерігається гуморальний дефіцит, а в 13 % — дефіцит IgA. Рецидивам аспіраційної пневмонії та бронхітів, синуситів (25–33 %) та отитів (4–7 %) сприяють краніофаціальні вади, що супроводжуються дисфагією та гастроезофагеальним рефлюксом. У дітей з уродженою вадою серця рецидиви респіраторної інфекції частіші [1, 10–21].
Основою імунної недостатності при синдромі делеції 22q11.2 є гіпоплазія тимуса та знижена продукція Т-лімфоцитів. Імунна недостатність варіює від нормального розвитку тимуса та продукування Т-клітин до відсутності Т-клітинної продукції. Цікаво, що прояви синдрому делеції 22q11.2 включають і порушення гуморальної імунної відповіді: гіпогаммаглобулінемію на першому році життя, дефіцит IgA та функціональні дефекти антитіл. Селективний дефіцит IgA може виявлятись у 10 % осіб з делецією і є особливо поширеним серед тих, хто має автоімунні захворювання [1, 10–21].
Автоімунне захворювання при синдромі 22q11.2DS є частим явищем. Ювенільний ревматоїдний артрит, часто поліартикулярний, виникає в 20–100 разів частіше, ніж у загальній популяції. Характерні ідіопатична тромбоцитопенія, гемолітична анемія, автоімунний тиреоїдит та інші автоімунні порушення. Ймовірно, у патогенезі автоімунних хвороб дефект Т-клітин діє синергічно з іншими факторами (наприклад, основним комплексом гістосумісності) [1, 10–21].
Краніофасціальні аномалії виявляють у 69 % пацієнтів із синдромом делеції 22q11.2 (табл. 2). Коротке тверде або м’яке піднебіння в комбінації з гіпотонією велофарингеальної мускулатури вважається характерною мальформацією. Лише 11 % пацієнтів із синдромом делеції 22q11.2 мають розщеплення твердого піднебіння від явного до непомітного дефекту, що діагностується тільки інструментально. В 1–2 % дітей має місце хейлосхізис (розщеплення губи). До додаткових черепно-лицьових особливостей, що можуть полегшити діагностику, відносять асиметрію обличчя під час посмішки або плачу (14 % пацієнтів із 22q11.2DS), аурикулярні аномалії з геміфаціальною мікросомією або без неї, краніосиностоз та характерні риси обличчя, такі як нависання повік, широке перенісся. Часто в дітей, яким спочатку діагностували делецію 22q11.2 через серцевий дефект, згодом виявлялася велофасціальна аномалія [1, 10–21].
/160-2.jpg)
Можливі аномалії вушної раковини (рис. 5), що включають періаурикулярні нарости, мікротію, анотію, звужений слуховий отвір; мальформації носа (широке перенісся, розширені ніздрі) [1].
У дітей із делецією 22q11.2 часто виявляють стридор у результаті судинного кільця, ларингомаляції, атрезії гортані та субглоткового стенозу; трахеоезофагеальну фістулу й езофагеальну атрезію. Нерідко діагностується сенсоневральне та кондуктивне зниження слуху [1, 10–21].
Ендокринні аномалії. Гіпокальціємія, як наслідок гіпопаратиреозу, виявляється в 17–60 % пацієнтів із синдромом делеції 22q11.2. Симптоми гіпокальціємії можуть включати тетанію, судоми, складність споживання їжі, стридор і втому. Гомеостаз кальцію з віком поступово нормалізується, хоча гіпокальціємія може і в подальшому розвиватися під час стресу (наприклад, під час хвороби, у період операції або в підлітковому віці). До можливих ендокринних розладів відносять гіпотиреоз, дефіцит гормону росту. Репродуктивна функція в жінок 22q11.2 за відсутності шизофренії або інтелектуальної недостатності не знижена. Репродуктивна здатність чоловіків з 22q11.2DS знижена [1, 10–21].
Затримка розвитку. Для дітей із синдромом делеції 22q11.2. характерна затримка фізичного та психомоторного розвитку. У грудному віці часто виявляється гіпотонія, ~50 % мають мікроцефалію, судоми, рідше має місце атаксія в результаті гіпоплазії мозочка, мультикістоз білої речовини мозку, гіпоплазія гіпоталамуса, полімікрогірія. Значну затримку росту (< 5-й перцентиль) мають 41 % дітей віком від 1 до 15 років, що обумовлено низькою концентрацією інсуліноподібного фактора росту та дефіцитом гормону росту в дітей з гіпоплазією гіпофіза. Пацієнти з делецією 22q11.2 часто демонструють значну затримку мовленнєвого розвитку: ~70 % дітей у віці 24 місяців не говорить або використовують лише кілька слів або знаків. Дві третини осіб потрапляють до діапазону IQ від 55 до 85 балів порівняно з еталонним діапазоном IQ у діапазоні 85–115 (середня: 100). Отже, труднощі в навчанні дуже часто зустрічаються в дошкільній та початковій школах, особливо в галузі математики та розуміння мови. Пацієнти після інсультів (наприклад, після зупинки серця, судом) або при аномалії розвитку головного мозку (наприклад, полімікрогірія) можуть мати гірший когнітивний прогноз [1, 10–21].
Психіатричні розлади. Особи з делецією 22q11.2DS мають підвищений ризик розвитку дефіциту уваги та розладів спектра аутизму, у 25 % виявляється шизофренія, що робить делецію 22q11.2 найвпливовішим молекулярно-генетичним фактором ризику шизофренії[1, 10–21].
Розлади харчування. Близько 36 % дітей з 22q11.2DS мають дисфагію, що не пов’язана з природженими вадами серця або аномаліями твердого піднебіння. Саме порушення координації ковтання й дихання у зв’язку з дизморфією й дисфункцією відповідних структур при онтогенезі третьої й четвертої зябрових дуг вважається причиною розладів харчування в цієї когорти дітей [1, 10–21].
Діагностика синдрому делеції 22q11.2 проводиться флуоресцентною гібридизацією in situ (методика недостатньо чутлива для виявлення делецій < 40 kb у межах 22q11.2) та більш чутливими методиками: мультиплексною лігазною ланцюговою реакцією (multiplex ligation-dependent probe amplification — MLPA) або за допомогою хромосомного мікрочіпу (chromosomal microarray — CMA) [23, 24].
Передімплантаційна генетична діагностика проводиться до переносу ембріона за умови підвищеного ризику хвороби в пробандів та при незбалансованій або збалансованій делеції з 22q11.2 хромосоми в батька. Пренатальне тестування вагітних за допомогою СМА рекомендоване жінкам із підвищеним ризиком розвитку хвороби на підставі сімейної історії та при підозрі мальформації в плода. Цільові методи, що включають, наприклад, FISH та MLPA, можуть бути використані для швидкої діагностики після аналізу CMA у вагітних та дітей всіх вікових груп при характерних аномаліях та затримці психічного й фізичного розвитку [25, 26].
Отже, якщо підозрюють мікроделецію 22q11.2 на підставі клінічних ознак, для швидкої діагностики можна застосовувати FISH. Однак CMA і MLPA мають вищу чутливість для виявлення нетипових делецій і дозволять визначити їх розмір.
Моніторинг дитини із синдромом делеції 22q11.2 здійснюється за принципом «система за системою»:
1. Він передбачає багатопрофільний командний підхід (педіатр, хірург, кардіоревматолог, кардіохірург, імунолог, ендокринолог, отоларинголог, клінічний генетик, алерголог, аудіолог, психолог, психіатр, стоматолог, гастроентеролог, пульмонолог, невролог, нейрохірург, офтальмолог, ортопед, пластичний хірург, уролог та логопед) [1, 27].
2. Педіатр виконує основну моніторингову та логістичну функцію, контролює фізичний, психічний розвиток дитини й соматичний статус, визначає рівень кальцію крові. Важлива модифікація годування дитини для запобігання аспірації їжі [1, 28].
3. Своєчасне виявлення природжених вад серця й судин та їх радикальну корекцію, постопераційне спостереження виконують кардіоревматолог, кардіохірург, хірург. До й після будь-яких операцій бажано дослідити рівень сироваткового кальцію, щоб своєчасно відкорегувати гіпокальціємію та уникнути гіпокальціємічних судом; а також кількість і функцію тромбоцитів для запобігання кровотечі. Перед хірургічними втручанням на шиї рекомендовано оцінити анатомію сонних артерій. При аденектомії — розглянути можливість порушення мовлення. Оцінити аномалії шийного відділу хребта перед інтубацією при хірургічному втручанні [1, 29].
4. Ендокринолог залучається за умови наявності гіпокальціємії, при затримці росту та клінічних ознаках гіпотиреозу. Рекомендоване визначення рівня іонізованого та неіонізованого кальцію в сироватці крові, рівнів соматотропного гормону, гормонів гіпофіза та щитоподібної залози. Газовані напої та споживання алкоголю можуть загострювати гіпокальціємію [1, 30, 33].
5. Оцінка тимічної функції виконується шляхом аналізу абсолютної кількості Т-клітин периферичної крові методом проточної цитометрії, що включає визначення рівня Т-клітин (CD3), наївних Т-клітин (як правило, CD4/CD45RA), Т-клітин пам’яті (як правило, CD4 або CD45RO), В-клітин (CD19) та натуральних кілерів (CD3–, CD56+, CD16+). Ультразвукова діагностика стану тимуса не є точною, оскільки розмір, відсутність або наявність тимічної тканини не прогнозує індивідуальну імунну функцію, хоча відсутність візуалізації тимуса є показанням для поглибленої диференціальної діагностики синдрому делеції 22q11.2. В осіб із рецидивуючою синопульмональною інфекцією часто виникають аномалії порушення відповіді антитіл на пневмококову полісахаридну вакцину. Імунолог вирішує питання імунізації дитини. У дітей із синдромом делеції 22q11.2 щеплення живими вакцинами не рекомендоване. Імунізацію неживими вакцинами дітям проводять за календарем імунізації з дослідженням антитіл для оцінки результатів імунізації. У випадку частих респіраторних інфекцій доцільне прийняття рішення щодо профілактичної антибактеріальної терапії, внутрішньовенного введення імуноглобулінів та трансплантації тимуса [1, 31, 32].
6. Рання діагностика та втручання психолога й психіатра покращують довгостроковий прогноз.
Отже, синдром делеції 22q11.2 хромосоми є найбільш поширеним синдромом мікроделеції з поліорганною дисфункцією. Частота синдрому делеції 22q11.2 коливається в межах від 1 : 3000 до 1 : 6000 новонароджених, проте Україні синдром діагностується рідко, що, на нашу думку, обумовлено відсутністю неонатального скринінгу й недостатньою обізнаністю лікарів щодо клінічних особливостей хвороби. Більшість новонароджених із синдромом 22q11.2 делеції мають природжену ваду серця, рідше — мальформації твердого піднебіння, затримку розвитку, шизофренію. Виявити типові й нетипові делеції на підставі клінічних ознак методами FISH, CMA і MLPA на сьогодні не становить складності, тому метою даної роботи було зосередити увагу на основних фенотипових появах хвороби для раннього виявлення синдрому делеції 22q11.2.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. McDonald-McGinn D.M. 22q11.2 deletion syndrome / E.S. Kathleen, B. Marino, N. Philip, A. Swillen, J.A.S. Vorstman, E.H. Zackai // Nat. Rev. Dis. Primers. — 2016. — № 1. — P. 1-45.
2. Grati F.R. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies / F.R. Grati // Prenat. Diagn. — 2015. — № 35. — P. 801-809.
3. Chien Y.H. Incidence of severe combined immunodeficiency through newborn screening in a Chinese population / Y.H. Chien / J. Formos. Med. Assoc. — 2015. — № 114. — P. 12-16.
4. Steinberg K.M. Single haplotype assembly of the human genome from a hydatidiform mole / K.M. Steinberg / Genome Res. — 2014. — № 24. — P. 2066-2076.
5. Chaisson M.J. Resolving the complexity of the human genome using single-molecule sequencing / M.J. Chaisson // Nature. — 2015. — № 517. — P. 608-611.
6. Ellegood J. Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion / J. Ellegood // Mol. Psychiatry. — 2014. — № 19. — P. 99-107.
7. Mukai J. Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia / J. Mukai // Neuron. — 2015. — № 86. — P. 680-695.
8. Earls L.R. A synaptic function approach to investigating complex psychiatric diseases / L.R. Earls, S.S. Zakharenko // Neuroscientist. — 2013. — № 20. — P. 257-271.
9. Karpinski B.A. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome / B.A. Karpinski // Dis. Model. Mech. — 2014. — № 7. — P. 245-257.
10. Pane L.S. Tbx1 is a negative modulator of Mef2c / L.S. Pane // Hum. Mol. Genet. — 2012. — № 21. — P. 2485-2496.
11. Diogo R. A new heart for a new head in vertebrate cardiopharyngeal evolution / R. Diogo // Nature. — 2015. — № 520. — P. 466-473.
12. Meechan D.W. Modeling a model: mouse genetics: 22q11.2 deletion syndrome, and disorders of cortical circuit development / D.W. Meechan // Prog. Neurobiol. — 2015. — № 130. — P. 1-28.
13. Xu B. Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion / B. Xu, P.K. Hsu, K.L. Stark, M. Karayiorgou, J.A. Gogos // Cell. — 2013. — № 152. — P. 262-275.
14. Milgrom-Hoffman M. Endothelial cells regulate neural crest and second heart field morphogenesis / M. Milgrom-Hoffman, I. Michailovici, N. Ferrara, E. Zelzer, E. Tzahor // Biol. Open. — 2014. — № 3. — P. 679-688.
15. Keyte A.L. Evolutionary and developmental origins of the cardiac neural crest: building a divided outflow tract / A.L. Keyte, M. Alonzo-Johnsen, M.R. Hutson // Birth Defects Res. C. Embryo Today. — 2014. — № 102. — P. 309-323.
16. Guna A. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms / A. Guna, N.J. Butcher, A.S. Bassett // J. Neurodev. Disord. — 2015. — № 7. — P. 18.
17. Zheng P. Molecular mechanisms of functional natural killer deficiency in patients with partial DiGeorge syndrome / P. Zheng // J. Allergy Clin. Immunol. — 2015. — № 135. — P. 1293-1302.
18. Paronett E.M. Ranbp1, deleted in DiGeorge/22q11.2 deletion syndrome, is a microcephaly gene that selectively disrupts layer 2/3 cortical projection neuron generation / E.M. Paronett, D.W. Meechan, B.A. Karpinski, A-S. LaMantia, T.M. Maynard // Cereb. Cortex. — 2014. — № 25. — P. 3977-3993.
19. Swillen A. Developmental trajectories in 22q11.2 deletion syndrome / A. Swillen, D. McDonald-McGinn // Am. J. Med. Genet. C. Semin. Med. Genet. — 2015. — № 169. — P. 172-181.
20. Fung W.L. Practical guidelines for managing adults with 22q11.2 deletion syndrome / W.L. Fung // Genet. Med. — 2015. — № 17. — P. 599-609.
21. Vergaelen E. 3 generation pedigree with paternal transmission of the 22q11.2 deletion syndrome: intrafamilial phenotypic variabi–lity / E. Vergaelen // Eur. J. Med. Genet. — 2015. — № 58. — P. 244-248.
22. Widdershoven J.C. A candidate gene approach to identify modifiers of the palatal phenotype in 22q11.2 deletion syndrome patients / J.C. Widdershoven // Int. J. Pediatr. Otorhinolaryngol. — 2013. — № 77. — P. 123-127.
23. Stransky C. Perioperative risk factors in patients with 22q11.2 deletion syndrome requiring surgery for velopharyngeal dysfunction / C. Stransky // Cleft Palate Craniofac. J. — 2015. — № 52. — P. 183-191.
24. Evers L.J. Psychopathology in adults with 22q11 deletion syndrome and moderate and severe intellectual disability / L.J. Evers // J. Intellect. Disabil. Res. — 2014. — № 58. — P. 915-925.
25. Vorstman J.A. Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome / J.A. Vorstman // JAMA Psychiatry. — 2015. — № 78. — P. 377-385.
26. Schneider M. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 deletion syndrome / M. Schneider // Am. J. Psychiatry. — 2014. — № 171. — P. 627-639.
27. Grewal J. Pregnancy in women with heart disease: risk assessment and management of heart failure / J. Grewal, C.K. Silversides, J.M. Colman // Heart Fail. Clin. — 2014. — № 10. — P. 117-129.
28. Mercer-Rosa L. 22q11.2 deletion syndrome is associated with perioperative outcome in tetralogy of Fallot / L. Mercer-Rosa, N. Pinto, W. Yang, R. Tanel, E. Goldmuntz // J. Thorac. Cardiovasc. Surg. — 2013. — № 146. — P. 868-873.
29. Hofstetter A.M. Live vaccine use and safety in DiGeorge syndrome / A.M. Hofstetter // Pediatrics. — 2014. — № 133. — P. 946-954.
30. Bjork A.H. Antibody deficiency in adults with 22q11.2 deletion syndrome / A.H. Bjork, S. Oskarsdottir, B.A. Andersson, V. Friman // Am. J. Med. Genet. A. — 2012. — № 158. — P. 1934-1940.
31. Basta M.N. A 35-year experience with syndromic cleft palate repair: operative outcomes and long-term speech function / M.N. Basta // Ann. Plast. Surg. — 2014. — № 73. — P. 130-135.
32. Wapner R.J. Expanding the scope of noninvasive prenatal tes–ting: detection of fetal microdeletion syndromes / R.J. Wapner // Am. J. Obstet. Gynecol. — 2015. — № 212. — P. 332.
33. Pretto D. Screening newborn blood spots for 22q11.2 deletion syndrome using multiplex droplet digital PCR / D. Pretto, D. Maar, C.M. Yrigollen, J. Regan, F. Tassone // Clin. Chem. — 2015. — № 61. — P. 182-190.

/156-1.jpg)
/157-1.jpg)
/160-1.jpg)