Роль клеточных реакций при пневмонии, вызванной
Staphylococcus aureus Эпителиоциты
Бактерии золотистого стафилококка, изменяя метаболизм и биосинтез нуклеотидов человеческих эпителиальных клеток как in vitro, так и in vivo, подавляют активность функционирования и нарушают структуру эпителия в респираторном тракте [29]. Бактериальные токсины и патогенассоци–ированные молекулярные структуры (PAMP) бактерий Staphylococcus aureus, взаимодействуя с образ-распознающими рецепторами (pattern recognition receptors — PRR) эпителиоцитов, обусловливают изменение транскрипции провоспалительных генов и повышение продукции цитокинов, хемокинов, антимикробных пептидов [9, 22]. В последнее время было установлено, что, кроме признанных PRR, в защите респираторного тракта от инфекционных патогенов участвуют рецепторы традиционных сенсорных путей трансдукции сигнала, а именно представители двух семейств хемосенсорных вкусовых рецепторов (taste receptor) — TAS1R, TAS2R (табл. 1) [50, 73].
Данные рецепторы, расположенные вне языковой области, в частности в респираторном тракте, участвуют в развитии противоинфекционного ответа [43]. Особую роль в противоинфекционной защите играют TAS2R. В организме человека идентифицировано около 25 изоформ TAS2R [45]. Так, активация TAS2R38, локализованного на апикальной мембране реснитчатых клеток эпителия синусов человека, молекулами кворум сенсинга — бактериальными ацил-гомосерин-лактонами (acyl-homoserine lactones — AHL), участвующими в формировании биопленки патогенных бактерий, индуцирует генерацию оксида азота [47]. Продемонстрировано, что бактерии Staphylococcus aureus, активируя TAS2R38 синоназального эпителия, вызывают генерацию оксида азота [13]. Активация одиночных хемосенсорных клеток через TAS2R инициирует увеличение внутриклеточной концентрации ионов кальция, что приводит к быстрой индукции синтеза и высвобождению антимикробных пептидов, в отличие от медленного действия активации TLR [14]. По всей вероятности, основной функцией рецепторов TAS2R в качестве сенсоров врожденной иммунной системы является предотвращение развития бактериальной биопленки.
С другой стороны, установлено, что горькие соединения (капсаицин) или бактериальные AHL способствуют высвобождению ацетилхолина, который стимулирует продукцию пептида, связанного с геном кальцитонина, и субстанции Р, что приводит к инициированию нейрогенного воспаления в ответ на бактериальную инвазию, в частности, в носовой полости [64].
Таким образом, вкусовые рецепторы респираторного тракта играют важную роль в реакции врожденной иммунной системы на внедрение инфекционного агента, вызывая развитие воспаления и продукцию антимикробных пептидов. Показано, что дисфункция или генетическая изменчивость рецепторов, участвующих в сенсинге горьких или сладких веществ, предопределяет развитие хронического стафилококкового синусита и респираторных инфекций у больных сахарным диабетом [63, 73]. Значение данных рецепторов как фактора риска развития или неблагоприятного течения пневмонии до настоящего времени не изучено.
Продукты жизнедеятельности бактерий Staphylococcus aureus нарушают барьерную функцию эпителия респираторного тракта, облегчая процесс инфицирования золотистым стафилококком [62].
Индукция секреции хемокинов эпителиальными клетками привлекает эффекторные иммунные клетки в очаг поражения легких и определяет развитие и характер воспалительного процесса. В защите от инфекционных патогенов участвуют разнообразные иммуноциты как врожденной, так и адаптивной иммунной системы (табл. 2).
/737-1.jpg)
Значимыми эпителиоцитарными хемокинами, рекрутирующими макрофаги, при стафилококковой инфекции респираторного тракта считаются гранулоцитарный колониестимулирующий (granulocyte colony stimulating factor — G-CSF) и гранулоцитарно-макрофагальный колониестимулирующий фактор (granulocyte-macrophage colony-stimulating factor — GM-CSF) [3, 76]. В ответ на инфицирование бактериями Staphylococcus aureus эпителиоциты продуцируют хемокин CCL2, который привлекает моноциты и макрофаги [21]. Накоп–ление Ly6ChiCD11b+-моноцитов в очаге заражения является определяющим моментом воспалительного процесса, индуцированного бактериальным патогеном. Данные клетки, получившие название «воспалительные моноциты», экспрессируют CCR2-хемокиновый рецептор, который способствует их эмиграции из костного мозга в периферическое русло крови. Нокаутные мыши Ccr2–/–, у которых Ly6Chi CD11b+-моноциты не могут выйти за пределы костного мозга, высоковосприимчивы к разнообразным бактериальным инфекциям [58, 67]. После рекрутирования в лимфоузлы воспалительные моноциты дифференцируются в DC и приобретают способность к презентации антигенов и продукции IL-12, что обусловливает развитие Th1-ответа иммунной системы [57].
Учитывая, что бактерии Staphylococcus aureus могут существовать как во внеклеточном пространстве, так и внутри клетки, макроорганизм обладает различными клеточными механизмами элиминации патогена [27]. Внеклеточные бактерии Staphylococcus aureus подвергаются киллингу преимущественно за счет неспецифических клеточных реакций: они поглощаются и разрушаются различными фагоцитами: альвеолярными макрофагами, моноцитами, дендритными клетками (DC), нейтрофилами. Однако эффективность процесса фагоцитоза зависит от предшествующего адаптивного ответа, в частности от связывания компонентов комплемента и специфических антител с поверхностью бактериальной стенки [69]. Также Т-клетки способствуют активности фагоцитоза, рекрутируя макрофаги и нейтрофилы из костного мозга в инфекционный очаг. В то же время элиминации бактерий Staphylococcus aureus, находящихся внутри фагосом, способствуют Т-клеточные цитокины, особенно IFN-γ [71]. Инфицированная клетка, в которой бактерии Staphylococcus aureus находятся в цитоплазме вне фагосом, лизируется цитотоксическими Т-клетками (cytotoxic T cells — CTL) или натуральными киллерами (natural killer — NK), что приводит к высвобождению патогена для повторного фагоцитоза [67].
Макрофаги и альвеолярные макрофаги
Респираторный тракт содержит многочисленные популяции резидентных фагоцитирующих клеток (HLA-DR+), которые участвуют в защите макроорганизма от патогенных бактерий. Альвеолярные макрофаги составляют около 93 % в субпопуляционной структуре макрофагов легочной ткани [52]. Помимо альвеолярных макрофагов (АМ), Vineet I. Patel и соавт. [60] были идентифицированы субпопуляции таких резидентных фагоцитирующих клеток, как лангерин+-, BDCA1–CD14+-, CD14+BDCA1+-, BDCA1+CD14–- и BDCA1–CD14-клетки. Клетки данных субпопуляций отличаются активностью интернализации различных бактерий, в том числе и золотистого стафилококка. Наиболее эффективная интернализация бактерий Staphylococcus aureus осуществляется АМ и CD14+-клетками. Необходимо отметить, что все резидентные фагоцитирующие клетки респираторного тракта более эффективно интернализируют бактерии Staphylococcus aureus, чем представителей Escherichia coli [60].
Альвеолярные макрофаги представляют резидентную медленно возобновляемую популяцию резистентных к конститутивному апоптозу провоспалительных клеток респираторного тракта и выступают в качестве клеточных компонентов первой линии антимикробной защиты, индуцируя воспаление и участвуя в процессе разрешения инфекционного процесса в легких. Несмотря на то, что фагоцитоз патогенных агентов, осуществляемый АМ, не столь эффективен, как нейтрофильный фагоцитоз, АМ играют исключительную роль в саногенезе стафилококковой пневмонии. Продемонстрировано, что транзиторное клодронатиндуцированное истощение АМ сопровождается чрезвычайной летальностью при индуцированной бактерией Staphylococcus aureus пневмонии у мышей, в то время как потеря DC практически не сопровождается увеличением уровня летальности [18, 54]. Показано, что АМ непосредственно интернализируют бактерии Staphylococcus aureus, используя макрофагальный рецептор, обладающий коллагеновой структурой (macrophage receptor with collagenous structure — MARCO). Предполагают, что именно данный скавенджерный рецептор играет центральную роль в макрофагопосредованной элиминации золотистого стафилококка [6].
В настоящее время различают девять субпопуляций макрофагов, в зависимости от фенотипа продуцирующих различного спектра цитокинов, хемокинов и поверхностных маркеров (табл. 3).
Основными макрофагальными субпопуляциями, которые принимают участие в воспалительной реакции, являются клетки с M1- и M2-фенотипом. Макрофаги отличаются высокой пластичностью: фенотип макрофага сильно зависит от цитокинового микроокружения клетки. Моноциты дифференцируются при наличии GM-CSF или макрофагального колониестимулирующего фактора (macrophage colony stimulating factor — M-CSF). Продемонстрировано, что моноциты при наличии GM-CSF и провоспалительных цитокинов (IFN-γ) или PAMP приобретают провоспалительный и бактерицидный фенотип M1, а при наличии M-CSF и IL-4 — М2, ассоциированный с ремоделированием и репарацией тканей [16, 78].
В свою очередь, М2-макрофаги состоят из трех субпопуляций: М2a, М2b, М2c. Установлено, что M2a-макрофаги индуцируются Th2-ассоциированными цитокинами (IL-4 и IL-13); М2b-макрофаги — LPS или сочетанием иммунных комплексов с IL-1β и М2c-макрофаги — IL-10, TGF-β или глюкокортикостероидами. M1-макрофаги характеризуются провоспалительным, а M2-макрофаги — противовоспалительным профилем цитокиновой продукции. Исключением являются М2b-макрофаги, которые активно продуцируют и провоспалительные цитокины, в том числе IL-1β, IL-6 [25, 38].
TLR2-ассоциированная активация транскрипционного фактора FOXO1 (forkhead box O1) в моноцитах, наблюдаемая при стафилококковой инфекции, способствует их дифференцировке в провоспалительные M1-макрофаги. Фактор транскрипции FOXO1 усиливает экспрессию цитокинов TNF-α, IL-1β, IL-6, хемокинов CCR2 и CCR7, которые способствуют миграции макрофагов, и CXCL10, привлекающего Т-клетки. В то же время при возбуждении стафилококковыми лигандами TLR2-ассоциированных сигнальных путей — PI3K/Akt и C-Raf/EK/ERK протеин FOXO1, расположенный внутриядерно, может быть фосфорилирован и экспортирован из ядра, что способствует M2-поляризации [72].
Взаимодействие и последующий фагоцитоз патогенных бактериальных агентов альвеолярными макрофагами зависят от экспрессии рецепторов иммуноглобулинов, комплемента, бета-глюкана, маннозы, а также нескольких типов скавенджерных рецепторов [75].
Все макрофаги, независимо от принадлежности к той или иной популяции, изначально обладают способностью фагоцитировать бактерии, обусловливая элиминацию Staphylococcus aureus. Также макрофаги участвуют в киллинге бактерий Staphylococcus aureus, генерируя активированные кислород-, азотсодержащие метаболиты (АКМ, ААМ) и антимикробные пептиды [20, 26].
Эффероцитоз как основной механизм физиологического снижения активности воспалительного процесса
Важнейшей функцией макрофагов является поглощение апоптотических иммуноцитов, обеспечивающее фагоцитарный клиренс апоптотических клеток — эффероцитоз (efferocytosis), активность которого определяет процесс реконвалесценции инфекционно-воспалительных заболеваний респираторного тракта [30]. Эффероцитоз, как правило, осуществляется профессиональными фагоцитами (макрофагами или DC), но может быть выполнен и с помощью непрофессиональных фагоцитов — эпителиальных клеток и фибробластов. Эффероцитоз обеспечивает быстрое удаление апоптотических клеток, предшествующее потере целостности их мембраны и высвобождению внутриклеточного содержимого [30, 36].
В развитии эффероцитоза различают три стадии: 1) высвобождение апоптотическими клетками хемотаксических веществ, привлекающих макрофаги; 2) хемотаксис макрофагов и их взаимодействие с апоптотической клеткой; 3) фагоцитоз и лизис апоптотических клеток. Основной противовоспалительный эффект эффероцитоза обусловлен физической секвестрацией погибающих клеток, что ограничивает высвобождение внутриклеточных молекулярных смерть-несущих структур, обладающих выраженной провоспалительной активностью (рис. 1) [23].
/742-1.jpg)
Миграцию макрофагов к апоптотическим клеткам индуцируют трифосфатнуклеотиды (аденин- и уридинтрифосфатнуклеотид — АТФ, УТФ соответственно), хемокин CX3CL1 и сигнальные липиды: лизофосфатидилхолин (lysophosphatidylcholine — lysoPC) и сфингозин-1-фосфат (sphingosine-phosphate — S1P) [4]. Апоптотические клетки высвобождают АТФ, УТФ через гексамерные паннексиновые каналы, активированные каспазой-3 и -7 (caspase-activated pannexin 1 — PANX1). Внеклеточно расположенные нуклеотиды АТФ, УТФ стимулируют рекрутирование фагоцитов, экспрессирующих пуринергические рецепторы P2Y. Хемокин CX3CL1 рекрутирует к апоптотической клетке CX3CR1+ фагоциты и функционирует в лимфоидной ткани и ткани головного мозга. S1P высвобождается из апоптотических клеток, обусловливая хемотаксис миелоидных клеток [23].
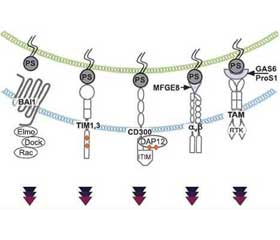
Макрофаги распознают апоптотические клетки по высокому уровню фосфатидилсерина (phosphatidylserine — PS) на поверхности их мембраны. В настоящее время идентифицировано не менее двенадцати рецепторов PS, участвующих в эффероцитозе, из которых наиболее изучены BAI (brain-specific angiogenesis inhibitor 1), TAM (Tyro3-Axl-Mer), TIM (T cell immunoglobulin domain), рецепторы стабилина и CD300 [61]. В отличие от циркулирующих моноцитов, DC и тканевых макрофагов альвеолярные макрофаги обладают многочисленными рецепторами, распознающими апоптотические клетки, и чрезвычайно высокой чувствительностью к апоптотическим клеткам в альвеолярном пространстве [55].
Апоптотические клетки после фагоцитирования содержатся в макрофаге в большой фагосоме, получившей название «эфферосома». Эфферосома способна объединяться с лизосомой и образовывать эфферолизосому, окончательно лизирующую апоптотическую клетку. В большинстве случаев фагоциты поглощают погибающую клетку целиком — именно так макрофаги поглощают апоптотические нейтрофилы [62]. Макрофаги, фагоцитировавшие апоптотические клетки, экспрессируют нуклеарный рецептор NR1H2 (nuclear receptor subfamily 1 group H member 2/LXRa, LXRb), рецепторы g, активируемые пероксисомными пролифераторами (peroxisome proliferator activated receptor g — PPARg), и ретиноидный X-рецептор α (retinoid X-receptor alpha — RXR-α), что способствует продукции TGF-β и IL-10, подавлению воспалительной реакции и репарации тканей [32].
Необходимо отметить, что эффероцитоз играет существенную роль и в элиминации бактерий, которые выживают во внутриклеточном пространстве нейтрофилов после фагоцитоза [53]. В частности, продемонстрировано, что бактерии Staphylococcus aureus способны выживать в нейтрофилах, тем самым защищая себя от клиренса из легочной ткани, выполняемого другими фагоцитами [34].
Согласно данным исследований мышиных моделей острого поражения легких, нарушения эффероцитоза в значительной мере предопределяют прогноз их исхода. В частности, показано, что инстилляция апоптотических клеток в респираторный тракт мышей после блеомицининдуцированного поражения легких снижает продукцию провоспалительных цитокинов, активность фиброзирования легких и приводит к увеличению уровня выживаемости животных [46]. Также внутрибрюшинное введение мышам экзогенного опсонина — фактора 8 эпидермального фактора роста, ассоциированного с глобулой молочного жира (milk fat globule-EGF factor 8 protein — MFG-E8), способствующего эффероцитозу, сопровождается снижением уровня продукции провоспалительных цитокинов в ткани легких, уменьшением объема поражения легких и уровня летальности экспериментальных мышей [19]. С другой стороны, при остром LPS-индуцированном поражении легких мышей с нокаутным геном Mfg-E8 наблюдаются высокая степень инфильтрации нейтрофилами очага поражения легких, более высокий уровень провоспалительных цитокинов и низкий уровень выживаемости экспериментальных животных [7]. Также введение TNF-α сопровождается подавлением активности клиренса апоптотических клеток из легких и, вероятно, ассоциированным с данным феноменом увеличением уровня воспаления легких [12].
Таким образом, активация эффероцитоза апоптотических нейтрофилов, предупреждая высвобождение провоспалительных цитокинов, способствует разрешению воспаления в респираторном тракте и репарации легочной ткани.
Установлено, что бактерии Staphylococcus aureus, используя различные механизмы, изменяют активность макрофагального эффероцитоза.
Однако во время стафилококковой пневмонии не отмечается изменение экспрессии молекул CD36, CD172 макрофагами в легочной ткани, от которых, как известно, зависит клиренс апопототических нейтрофилов [17]. Taylor S. Cohen [17] продемонстрировал, что фактор вирулентности токсин-α Staphylococcus aureus изменяет экспрессию и секрецию протеина 61, богатого цистеином ангиогенного индуктора (cysteine rich angiogenic inducer 61 — CYR61), эпителиальными клетками и V-set иммунорегуляторного рецептора (V-set immunoregulatory receptor — VSIR/death domain1α — DD1α) макрофагами. Протеин CYR61 связывается одновременно с фосфатидилсерином на поверхности апоптотических клеток и с интегринами (V3/V5) макрофагов и других клеток (табл. 4). В последующем, активируя Rho-ассоциированный сигнальный путь в макрофагах, CYR61 инициирует фагоцитоз. В отличие от CYR61 участие VSIR в эффероцитозе не зависит от фосфатидилсерина. Рецептор VSIR экспрессируется как апоптотическими клетками, так и макрофагами, и поглощение апоптотической клетки инициируется VSIR-VSIR-связыванием. Согласно полученным результатам, авторы считают, что бактерии Staphylococcus aureus, используя токсин-α, ингибируют экспрессию CYR61 и VSIR в ткани легкого, что обусловливает снижение активности клиренса апоптотических нейтрофилов. Необходимо отметить, что CYR61 и VSIR оказывают влияние не только на эффероцитоз. Так, гиперэкспрессия Cyr61 способствует развитию воспаления и деструкции ткани легкого, реализуя свое действие через взаимодействие с различными рецепторами иммуноцитов [31].
/741-1.jpg)
Протеин CYR61 индуцирует транскрипционные изменения, характерные для M1-макрофагов, в том числе увеличивает экспрессию TNF-α, IL-1α, IL-1β, IL-6, IL-12, CCL3, CCL7 и CXCL10 NF-kB-зависимым образом [8].
В отличие от CYR61 VSIR является мощным ингибитором Т-клеточной активации, а его дефицит приводит к развитию аутоиммунного фенотипа [74].
Mallary C. Greenlee-Wacker и соавт. [33] установили, что бактерии MRSA способствуют экспрессии молекулы CD47, которая представляет собой компонент сигнальной системы «не ешь меня», и инфицированные нейтрофилы не фагоцитируются или неэффективно поглощаются макрофагами. Кроме того, бактерии MRSA индуцируют изменение макрофагального фенотипа: макрофаги, инфицированные MRSA, продуцируют больше хемокин IL-8/СХСL8, чем цитокины IL-1β и TNF-α. Бактерии MRSA вызывают лизис фагоцитирующих клеток через 6 часов после фагоцитоза без активации каспазы. Лизис клеток зависит от рецепторвзаимодействующего протеина 1, в связи с чем авторы полагают, что бактерии MRSA, вероятно, ингибируя механизмы эффероцитоза макрофагов, индуцируют запрограммированный некроз или некроптоз нейтрофилов.
Таким образом, стафилококковые бактерии, особенно MRSA, для обеспечения собственного выживания подавляют эффероцитоз, что обусловливает неблагоприятное течение заболевания.
Staphylococcus aureus-индуцированная гибель фагоцитов как регулятор процесса воспаления
Клетки макроорганизма в ответ на инфицирование могут реагировать развитием собственной гибели, способствуя элиминации патогена. С другой стороны, бактериальные патогены способны непосредственно ингибировать активность ответа иммунной системы, избирательно индуцируя гибель некоторых иммунных клеток, в частности макрофагов, подавляя активность воспалительного процесса [11, 56].
Во время стафилококковой инфекции Staphylococcus aureus-индуцированная гибель фагоцитов, активность которой определяет течение воспалительного процесса, может быть представлена такими программируемыми процессами, как пироптоз, апоптоз, некроптоз. Развитие пироптоза обусловлено возбуждением образ-распознающих рецепторов и функционированием инфламмасом с активацией каспаз, апоптоза и некроптоза активацией TNFR1 и серинтреониновой протеинкиназы 1 (serine/threonine-protein kinase 1 — RIPK1). Возбуждение каспазы-8 в ответ на проапоптотический стимул приводит к развитию апоптоза, а ингибирование каспазы-8 — некроптоза макрофагов. Однако Oliver Kepp и соавт. [40] считают, что пироптоз представляет собой вариант апоптоза или некроптоза. Апоптотическая гибель характеризуется потерей клеткой внутриклеточной воды, распадом клетки на апоптотические тельца и поглощением их макрофагами без высвобождения биологически активных веществ. Некроптоз отличается развитием клеточного отека (онко–птоза), разрыва клеточной мембраны с высвобождением внутриклеточных DAMP, инициирующих неспецифические механизмы иммунной системы. Лигирование предварительно собранного TNFR1 триммера приводит к изменению конформации, которая способствует формированию мультимолекулярного образования Complex I. Данный мембранный комплекс состоит из TRADD, TRAF2, RIPK1, cIAP1, cIAP2 и комплекса линейных убиквитиновых цепей (linear ubiquitin chain assembly Complex — LUBAC). Убиквитиновая сеть Complex I рекрутирует и активирует ингибитор киназы кВ (inhibitor of κB kinase — IKK). Активированный IKK фосфорилирует IκBα, что приводит к ее протеасомной деградации и ядерной транслокации димеров NF-kB в ядро клетки, что вызывает экспрессию генов, участвующих в воспалении и способствующих выживанию клетки. NF-kB-зависимая индукция cFLIPL ингибирует развитие как апоптоза, так и некроптоза [15].
Клеточная гибель, индуцирующая воспаление
Пироптоз
Пироптоз, как форма запрограммированной гибели клеток, представляет собой особый эффекторный механизм врожденного иммунитета у позвоночных животных [39]. В течение долгого времени под пироптозом понималась каспаза-1-опосредованная гибель моноцитов [10]. Однако было установлено, что в развитии пироптоза участвуют мышиная каспаза-11 и человеческие каспаза-4 и -5, чувствительные к внутриклеточному липополисахариду. Каспаза-1, мышиная каспаза-1 и человеческие каспаза-4, -5, -11 взаимодействуют со своим лигандом гасдермином D (gasdermin D — GSDMD), который является пироптотическим «палачом» клетки. –После отщепления от GSDMD при помощи каспазы его N-терминальный фрагмент, переместившись к внутренней поверхности цитоплазматической мембраны, встраивается в нее и формирует поры, через которые высвобождаются активные провоспалительные цитокины IL-1β и IL-18 [66]. Через гасдерминовые поры во внутриклеточный континуум клетки проникает вода, способствуя развитию внутриклеточного отека и нарушению целостности мембраны макрофага, что приводит к гибели клетки (рис. 2) [28, 49].
/743-1.jpg)
Золотистый стафилококк индуцирует инфламмасомы и пироптоз резидентных макрофагов, причем уровень ответной реакции зависит от фенотипа макрофагов. Так, Solene Accarias и соавт. [1] показали, что мышиные и человеческие М1-макрофаги после инфицирования золотистым стафилококком реагируют достоверно более высоким увеличением активности каспазы-1 в сочетании с более высоким уровнем высвобождения IL-1 и потерей целостности клеточной мембраны, чем М2-макрофаги. Исследуя течение стафилококковой инфекции у мышей двух линий: C57BL/6 и DBA/2, резистентных и восприимчивых к инфекции соответственно, авторы продемонстрировали, что данные линии мышей отличаются уровнем инфламмасома-зависимого Staphylococcus aureus-индуцированного пироптоза макрофагов. У резистентных к Staphylococcus aureus C57BL/6 мышей наблюдается высокий уровень пироптоза макрофагов, который сопровождается высвобождением значительного количества IL-1β в сочетании с рестриктированной секрецией NF-kB-зависимых провоспалительных цитокинов (TNF-a, IL-6) и хемокинов (CXCL1 и CXCL2). И наоборот, у восприимчивых к Staphylococcus aureus DBA/2 мышей отмечается высокий уровень выживания макрофагов в сочетании с массивным высвобождением TNF-a, IL-6, CXCL1 и CXCL2. Назначение инфламмасомных индукторов (нигерицина и АТФ) DBA/2 мышам приводит к повышению секреции IL-1, усилению процесса пироптоза макрофагов и ингибированию продукции NF-kB-зависимых провоспалительных цитокинов.
Клеточная гибель, ингибирующая воспаление
Активация TNFR1 и киназ RIPK1, RIPK3 в зависимости от активности каспазы-8 может привести к развитию некроптоза и апоптоза. Инактивация каспазы-8 сопровождается сохранением полных молекулярных форм киназ RIPK1, RIPK3 и развитием некроптоза, а ее активация — расщеплением киназ RIPK1, RIPK3 и выживанием или апоптозом клетки. Формирование мультимолекулярного образования, состоящего из расщепленных киназ RIPK1, RIPK3 и димера каспазы-8, сопровождается развитием апоптоза, а комплекса, состоящего из расщепленных киназ RIPK1, RIPK3, каспазы 8 и регулятора апоптоза cFLIPL/CFLAR (CASP8 and FADD like apoptosis regulator), — выживанием клетки [2].
Некроптоз
Некроптоз в патогенезе пневмонии играет несколько ролей. Активация некроптоза может привести к сокращению представительства некоторых типов иммунных клеток, особенно макрофагов, и тем самым способствовать развитию инфекционного процесса. В то же время некроптоз способствует элиминации патогенных микроорганизмов, ликвидации зараженных клеток и подавлению чрезмерной активности воспалительного процесса [2, 59]. Некроптоз индуцируется при возбуждении рецептора смерти суперсемейства TNF, в частности TNFR1, TLR3 и TLR4, а также рецепторов IFN-γ [68]. Интернализация рецептора TNFR1 сопровождается его диссоциацией с RIPK1 и последующим формированием мультимолекулярного образования Complex IIa, содержащего RIPK1, RIPK3, FADD (Fas associated via death domain), прокаспазу-8 и cFLIPL/CFLAR. Димеризация каспазы-8 с длинной формой cFLIPL/CFLAR ингибирует полную активацию каспазы-8, что предотвращает развитие апоптоза клетки. После деградации cFLIPL/CFLAR, фосфорилирования киназы RIPK1 происходят рекрутирование киназы RIPK3 и взаимодействие RIPK-киназ, осуществляемое с использованием гомотипичного мотива RIP (RIP homotypic interaction motif). Данное взаимодействие приводит к формированию амилоидоподобного образования Complex IIc (RIPK1, RIPK3) или некросомы, которая рекрутирует и активирует псевдокиназу смешанной линии (mixed lineage kinase domain-like — MLKL). Киназа RIPK3 фосфорилирует аминокислотные остатки Thr357 и Ser358 псевдокиназы MLKL, вызывая олигомеризацию ее молекул и транслокацию олигомера MLKL к внутренней поверхности цитоплазматической мембраны. В свою очередь, олигомер MLKL индуцирует нарушение целостности мембраны и, как следствие, гибель клетки [15, 56, 70].
Бактерии Staphylococcus aureus могут активировать RIPK1/RIPK3-сигнальный путь, возбуждая TNFR1 и индуцируя продукцию IFN-γ [2].
Некроптоз, индуцируемый бактериями Staphylococcus aureus, является высокотоксинзависимой реакцией, триггерами которой выступают α-гемолизин, фенолсолютабные модулины и лейкоцидин LukAB [42]. Kipyegon Kitur и соавт. [41] показали, что некроптоз фагоцитов, опосредованный возбуждением RIPK1/RIPK3/MLKL-пути, рестриктирует активность воспаления. Неспособность активировать некроптоз и ассоциированное с ним высвобождение IL-1β сопровождается ослаб–лением стафилококкового клиренса. Также продемонстрировано, что стафилококковая инфекция у нокаутных мышей Mlkl–/– и Ripk3–/– характеризуется сочетанием высокого уровня бактериальной нагрузки и чрезмерным воспалением за счет избытка продукции IL-1β. Анализ клиренса бактерий Staphylococcus aureus при инфицировании мышей позволил установить, что в ткани легкого у мышей Ripk3–/– представительство макрофагов, экспрессирующих противовоспалительные молекулы CD200R и CD206, значительно выше, чем у мышей дикого типа. Данный тип АМ обычно ингибирует воспалительную реакцию в дыхательных путях, и их высокий уровень выживания в легких мышей Ripk3–/–, вероятно, является одной из основных причин благоприятного течения стафилококковой пневмонии. Применение ингибиторов некроптоза (некростатина-1s, ингибитора RIPK1) также приводит к увеличению выживаемости АМ [42].
Апоптоз
Золотистый стафилококк способен активировать не только RIPK1/RIPK3/MLKL, каспаза-1-ассоциированные сигнальные пути, но и каспазу-8 [37]. Тем не менее апоптотический путь гибели фагоцитов непосредственно не участвует в клиренсе стафилококка, а также в контроле продукции IL-1β [41]. Вместе с тем установлено, что стафилококковый энтеротоксин B (staphylococcal enterotoxin B) вызывает апоптоз ТНР-1-клеток, подавляя активность воспалительного процесса [77].
Развитие пироптоза, апоптоза и некроптоза при пневмонии, вызванной Staphylococcus aureus, схематично представлено на рис. 3.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Accarias S, Lugo-Villarino G, Foucras G, Neyrolles O, Boullier S, Tabouret G, et al. Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. Eur J Immunol. 2015 Mar;45(3):794-806. doi: 10.1002/eji.201445098.
2. Ahn D, Prince A. Participation of Necroptosis in the Host Response to Acute Bacterial Pneumonia. J Innate Immun. 2017;9(3):262-270. doi: 10.1159/000455100. Epub 2017 Jan 27.
3. Alp E, Gozukucuk S, Canoz O, Kirmaci B, Doganay M. Effect of granulocyte colony-stimulating factor in experimental methicillin resistant Staphylococcus aureus sepsis. BMC Infect Dis. 2004 Oct 18;4:43. doi: 10.1186/1471-2334-4-43.
4. Angsana J, Chen J, Liu L, Haller CA, Chaikof EL. Efferocytosis as a regulator of macrophage chemokine receptor expression and polarization. Eur J Immunol. Jul;46(7):1592-9. doi: 10.1002/eji.201546262.
5. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014 Oct 7;5:491. doi: 10.3389/fimmu.2014.00491. eCollection 2014.
6. Arredouani MS, Palecanda A, Koziel H, et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol. 2005 Nov 1;175(9):6058-64. doi: 10.4049/jimmunol.175.9.6058.
7. Aziz M, Matsuda A, Yang WL, Jacob A, Wang P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol. 2012 Jul 1;189(1):393-402. doi: 10.4049/jimmunol.1200262.
8. Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol. 2010 Mar 15;184(6):3223-32. doi: 10.4049/jimmunol.0902792.
9. Bekeredjian-Ding I, Stein C, Uebele J. The Innate Immune Response Against Staphylococcus aureus. In: Current Topics in Microbiology and Immunology. Berlin: Springer; 2015 Dec 15. 1-34 pp. doi: 10.1007/82_2015_5004.
10. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009 Feb;7(2):99-109. doi: 10.1038/nrmicro2070.
11. Blériot C, Lecuit M. The interplay between regulated necrosis and bacterial infection. Cell Mol Life Sci. 2016 Jun;73(11-12):2369-78. doi: 10.1007/s00018-016-2206-1.
12. Borges VM, Vandivier RW, McPhillips KA, et al. TNFalpha inhibits apoptotic cell clearance in the lung, exacerbating acute inflammation. Am J Physiol Lung Cell Mol Physiol. 2009 Oct;297(4):L586-95. doi: 10.1152/ajplung.90569.2008.
13. Carey RM, Chen B, Adappa ND, et al. Human upper airway epithelium produces nitric oxide in response to Staphylococcus epidermidis. Int Forum Allergy Rhinol. 2016 Dec;6(12):1238-1244. doi: 10.1002/alr.21837.
14. Carey RM, Lee RJ, Cohen NA. Taste Receptors in Upper Airway Immunity. Adv Otorhinolaryngol. 2016;79:91-102. doi: 10.1159/000445137.
15. Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79-106. doi: 10.1146/annurev-immunol-032414-112248.
16. Chung S, Ranjan R, Lee YG, et al. Distinct role of FoxO1 in M-CSF- and GM-CSF-differentiated macrophages contributes LPS-mediated IL-10: implication in hyperglycemia. J Leukoc Biol. 2015 Feb;97(2):327-39. doi: 10.1189/jlb.3A0514-251R.
17. Cohen TS, Jones-Nelson O, Hotz M, et al. S. aureus blocks efferocytosis of neutrophils by macrophages through the activity of its virulence factor alpha toxin. Sci Rep. 2016 Oct 14;6:35466. doi: 10.1038/srep35466.
18. Cole J, Aberdein J, Jubrail J, Dockrell DH. The role of macrophages in the innate immune response to Streptococcus pneumoniae and Staphylococcus aureus: mechanisms and contrasts. Adv Microb Physiol. 2014;65:125-202. doi: 10.1016/bs.ampbs.2014.08.004.
19. Cui T, Miksa M, Wu R, et al. Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am J Respir Crit Care Med. 2010 Feb 1;181(3):238-46. doi: 10.1164/rccm.200804-625OC.
20. de Morais NG, da Costa TB, Pedrosa AL, et al. Effect of neonatal malnutrition on expression of nitric oxide synthase enzyme, production of free radicals and in vitro viability of alveolar macrophages infected with methicillin-sensitive and methicillin-resistant Staphylococcus aureus. Eur J Nutr. 2016 Feb;55(1):403-11. doi: 10.1007/s00394-015-0861-x.
21. Desouza IA, Franco-Penteado CF, Camargo EA, et al. Inflammatory mechanisms underlying the rat pulmonary neutrophil influx induced by airway exposure to staphylococcal enterotoxin type A. Br J Pharmacol. 2005 Nov;146(6):781-91. doi: 10.1038/sj.bjp.0706393.
22. Eiffler I, Behnke J, Ziesemer S, et al. Staphylococcus aureus α-toxin-mediated cation entry depolarizes membrane potential and activates p38 MAP kinase in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2016 Sep 1;311(3):L676-85. doi: 10.1152/ajplung.00090.2016.
23. Elliott MR, Koster KM, Murphy PS. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J Immunol. 2017 Feb 15;198(4):1387-1394. doi: 10.4049/jimmunol.1601520.
24. Emre Y, Imhof BA. Matricellular protein CCN1/CYR61: a new player in inflammation and leukocyte trafficking. Semin Immunopathol. 2014 Mar;36(2):253-9. doi: 10.1007/s00281-014-0420-1.
25. Ferrante CJ, Leibovich SJ. Regulation of Macrophage Polarization and Wound Healing. Adv Wound Care (New Rochelle). 2012 Feb;1(1):10-16. doi: 10.1089/wound.2011.0307.
26. Flannagan RS, Heit B, Heinrichs DE, et al. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens. 2015 Nov 27;4(4):826-68. doi: 10.3390/pathogens4040826.
27. Fraunholz M, Sinha B. Intracellular Staphylococcus aureus: live-in and let die. Front Cell Infect Microbiol. 2012 Apr 24;2:43. doi: 10.3389/fcimb.2012.00043.
28. Gaidt MM, Hornung V. Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J. 2016 Oct 17;35(20):2167-2169. doi: 10.15252/embj.201695415.
29. Gierok P, Harms M, Methling K, Hochgräfe F, Lalk M. Staphylococcus aureus Infection Reduces Nutrition Uptake and Nucleotide Biosynthesis in a Human Airway Epithelial Cell Line. Metabolites. 2016 Nov 9;6(4). pii: E41. doi: 10.3390/metabo6040041.
30. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. 2016 Jul;38(4):409-23. doi: 10.1007/s00281-016-0555-3.
31. Grazioli S, Gil S, An D, et al. CYR61 (CCN1) overexpression induces lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2015 Apr 15;308(8):L759-65. doi: 10.1152/ajplung.00190.2014.
32. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016 Jun;23(6):915-26. doi: 10.1038/cdd.2015.172.
33. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol. 2014 May 15;192(10):4709-17. doi: 10.4049/jimmunol.1302692.
34. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000 Apr 1;164(7):3713-22. doi: 10.4049/jimmunol.164.7.3713.
35. Hermann I, Räth S, Ziesemer S, et al. Staphylococcus aureus hemolysin A disrupts cell-matrix adhesions in human airway epithelial cells. Am J Respir Cell Mol Biol. 2015 Jan;52(1):14-24. doi: 10.1165/rcmb.2014-0082OC.
36. Hodge SJ, Tran H, Hamon R, et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am J Physiol Lung Cell Mol Physiol. 2017 May 1;312(5):L678-L687. doi: 10.1152/ajplung.00518.2016.
37. Hu Q, Cui X, Tao L, Xiu L, Wang T, Wang X. Staphylococcus aureus induces apoptosis in primary bovine mammary epithelial cells through Fas-FADD death receptor-linked caspase-8 signaling. DNA Cell Biol. 2014 Jun;33(6):388-97. doi: 10.1089/dna.2013.2195.
38. Jiang Z, Zhu L. Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy. 2016 Jun 3;9:101-7. doi: 10.2147/JAA.S104508. eCollection 2016.
39. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015 May;265(1):130-42. doi: 10.1111/imr.12287.
40. Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis - a cell death modality of its kind? Eur J Immunol. 2010 Mar;40(3):627-30. doi: 10.1002/eji.200940160.
41. Kitur K, Wachtel S, Brown A, et al. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016 Aug 23;16(8):2219-30. doi: 10.1016/j.celrep.2016.07.039.
42. Kitur K, Parker D, Nieto P, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015 Apr 16;11(4):e1004820. doi: 10.1371/journal.ppat.1004820.
43. Lee RJ, Cohen NA. Bitter and sweet taste receptors in the respiratory epithelium in health and disease. J Mol Med (Berl). 2014 Dec;92(12):1235-44. doi: 10.1007/s00109-014-1222-6.
44. Lee RJ, Cohen NA. Taste receptors in innate immunity. Cell Mol Life Sci. 2015 Jan;72(2):217-36. doi: 10.1007/s00018-014-1736-7.
45. Lee RJ, Cohen NA. The emerging role of the bitter taste receptor T2R38 in upper respiratory infection and chronic rhinosinusitis. Am J Rhinol Allergy. 2013 Jul-Aug;27(4):283-6. doi: 10.2500/ajra.2013.27.3911.
46. Lee YJ, Moon C, Lee SH. Apoptotic cell instillation after bleomycin attenuates lung injury through hepatocyte growth factor induction. Eur Respir J. 2012 Aug;40(2):424-35. doi: 10.1183/09031936.00096711.
47. Li F, Zhou M. Depletion of bitter taste transduction leads to massive spermatid loss in transgenic mice. Mol Hum Reprod. 2012 Jun;18(6):289-97. doi: 10.1093/molehr/gas005.
48. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017 Jan 5;117(1):7-18. doi: 10.1160/TH16-08-0593.
49. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016 Jul 7;535(7610):153-8. doi: 10.1038/nature18629.
50. Lu P, Zhang CH, Lifshitz LM, ZhuGe R. Extraoral bitter taste receptors in health and disease. J Gen Physiol. 2017 Feb;149(2):181-197. doi: 10.1085/jgp.201611637.
51. Marcenaro E, Carlomagno S, Pesce S, et al. Bridging innate NK cell functions with adaptive immunity. Adv Exp Med Biol. 2011;780:45-55. doi: 10.1007/978-1-4419-5632-3_5.
52. Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007 Dec;33(10):493-505. doi: 10.1080/01902140701756562.
53. Martin CJ, Peters KN, Behar SM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol. 2014 Feb;17:17-23. doi: 10.1016/j.mib.2013.10.007.
54. Martin FJ, Parker D, Harfenist BS, Soong G, Prince A. Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect Immun. 2011 May;79(5):1898-904. doi: 10.1128/IAI.01299-10.
55. McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest. 2013 Jun;143(6):1750-7. doi: 10.1378/chest.12-2413.
56. Moriwaki K, Chan FK. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016 Jun;73(11-12):2325-34. doi: 10.1007/s00018-016-2203-4.
57. Nakano H, Lin KL, Yanagita M, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol. 2009 Apr;10(4):394-402. doi: 10.1038/ni.1707.
58. Nombela-Arrieta C, Isringhausen S. The Role of the Bone Marrow Stromal Compartment in the Hematopoietic Response to Microbial Infections. Front Immunol. 2017 Jan 20;7:689. doi: 10.3389/fimmu.2016.00689. eCollection 2016.
59. Parker D, Prince A. Immunoregulatory effects of necroptosis in bacterial infections. Cytokine. 2016 Dec;88:274-275. doi: 10.1016/j.cyto.2016.09.024.
60. Patel VI, Booth JL, Duggan ES, et al. Transcriptional Classification and Functional Characterization of Human Airway Macrophage and Dendritic Cell Subsets. J Immunol. 2017 Feb 1;198(3):1183-1201. doi: 10.4049/jimmunol.1600777.
61. Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev. 2016 Jan;269(1):44-59. doi: 10.1111/imr.12376.
62. Robb CT, Regan KH, Dorward DA, Rossi AG. Key mechanisms governing resolution of lung inflammation. Semin Immunopathol. 2016 Jul;38(4):425-48. doi: 10.1007/s00281-016-0560-6.
63. Rom DI, Christensen JM, Alvarado R, Sacks R, Harvey RJ. The impact of bitter taste receptor genetics on culturable bacteria in chronic rhinosinusitis. Rhinology. 2017 Feb 19. doi: 10.4193/Rhin16.181.
64. Saunders CJ, Christensen M, Finger TE, Tizzano M. Cholinergic neurotransmission links solitary chemosensory cells to nasal inflammation. Proc Natl Acad Sci U S A. 2014 Apr 22;111(16):6075-80. doi: 10.1073/pnas.1402251111.
65. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421-52. doi: 10.1146/annurev.immunol.26.021607.090326.
66. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017 Apr;42(4):245-254. doi: 10.1016/j.tibs.2016.10.004.
67. Taylor AL, Cross EL, Llewelyn MJ. Induction of contact-dependent CD8(+) regulatory T cells through stimulation with staphylococcal and streptococcal superantigens. Immunology. 2012 Feb;135(2):158-67. doi: 10.1111/j.1365-2567.2011.03529.x.
68. Thapa RJ, Nogusa S, Chen P, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):E3109-18. doi: 10.1073/pnas.1301218110.
69. van Kessel KP, Bestebroer J, van Strijp JA. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front Immunol. 2014 Sep 26;5:467. doi: 10.3389/fimmu.2014.00467.
70. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016 Apr 1;352(6281):aaf2154. doi: 10.1126/science.aaf2154.
71. Wang XY, Huang ZX, Chen YG, et al. A Multiple Antigenic Peptide Mimicking Peptidoglycan Induced T Cell Responses to Protect Mice from Systemic Infection with Staphylococcus aureus. PLoS One. 2015 Aug 28;10(8):e0136888. doi: 10.1371/journal.pone.0136888.
72. Wang YC, Ma HD, Yin XY, et al. Forkhead Box O1 Regulates Macrophage Polarization Following Staphylococcus aureus Infection: Experimental Murine Data and Review of the Literature. Clin Rev Allergy Immunol. 2016 Dec;51(3):353-369. doi: 10.1007/s12016-016-8531-1.
73. Workman AD, Palmer JN, Adappa ND, Cohen NA. The Role of Bitter and Sweet Taste Receptors in Upper Airway Immunity. Curr Allergy Asthma Rep. 2015 Dec;15(12):72. doi: 10.1007/s11882-015-0571-8.
74. Yoon KW, Byun S, Kwon E, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015 Jul 31;349(6247):1261669. doi: 10.1126/science.1261669.
75. Yu X, Guo C, Fisher PB, et al. Scavenger Receptors: Emerging Roles in Cancer Biology and Immunology. Adv Cancer Res. 2015;128:309-64. doi: 10.1016/bs.acr.2015.04.004.
76. Zbinden C, Stephan R, Johler S, et al. The inflammatory response of primary bovine mammary epithelial cells to Staphylococcus aureus strains is linked to the bacterial phenotype. PLoS One. 2014 Jan 30;9(1):e87374. doi: 10.1371/journal.pone.0087374.
77. Zhang X, Shang W, Yuan J, et al. Positive Feedback Cycle of TNFα Promotes Staphylococcal Enterotoxin B-Induced THP-1 Cell Apoptosis. Front Cell Infect Microbiol. 2016 Sep 21;6:109. doi: 10.3389/fcimb.2016.00109. eCollection 2016.
78. Zhou D, Huang C, Lin Z, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014 Feb;26(2):192-7. doi: 10.1016/j.cellsig.2013.11.004.

/737-1.jpg)
/739-1.jpg)
/742-1.jpg)
/741-1.jpg)
/743-1.jpg)
/744-1.jpg)