Вступ
Неінфекційні захворювання (НІЗ) визнані Всесвітньою організацією охорони здоров’я (ВООЗ) однією з основних проблем XXI століття, а профілактика і боротьба з ними є пріоритетними напрямками. Серед усіх факторів ризику НІЗ ожиріння має найбільшу питому вагу. Поширеність дитячого і підліткового ожиріння зростає в усьому світі, створюючи загрозу здоров’ю та якості життя дітей. Ожиріння є безпосередньою причиною захворювань шлунково-кишкового тракту, опорно-рухового апарату, апное під час сну, а також раннього розвитку серцево-судинних захворювань і цукрового діабету 2 типу [1–3].
За оцінками ВООЗ (2013), у 42 млн дітей віком до 5 років спостерігається надмірна вага або ожиріння [1]. У США 15,8 % дітей у віці від 6 до 11 років і 16,1 % підлітків мають підвищений індекс маси тіла (ІМТ) [4]. Аналогічні тенденції спостерігаються і в більшості європейських країн, де надмірна вага і ожиріння, що визначені відповідно до критеріїв Міжнародної цільової групи, наявні в 31,8 % дітей шкільного віку [5].
Дані Bogalusa Heart Study продемонстрували, що майже 20 % дітей, які страждають від ожиріння, мають принаймні один значимий кардіоваскулярний фактор ризику (гіперхолестеринемія, гіперінсулінемія, гіпертригліцеридемія, гіпертонія), наявність множинних факторів ризику тісно пов’язана з раннім розвитком атеросклерозу [6]. З урахуванням того, що серцево-судинні захворювання є першою причиною захворюваності та смертності в дорослому віці, епідемія дитячого ожиріння становить собою реальний тягар для популяції загалом [6, 7]. Наявність надлишкової маси тіла може призводити до вісцерального ожиріння й ектопічного накопичення жиру з ураженням печінки, біліарного тракту, скелетної мускулатури, серця, а також підшлункової залози (ПЗ) (із розвитком стеатозу органа) [8]. Відомо, що надмірне накопичення жирової тканини в печінці може призводити до цитокінопосередкованого запалення, окислювального стресу з подальшим можливим розвитком запалення в органі — стеатогепатиту. Неалкогольний стеатогепатит тісно пов’язаний із компонентами метаболічного синдрому, зокрема резистентністю до інсуліну, майже у всіх пацієнтів із неалкогольним стеатогепатитом спостерігаються субклінічна резистентність до інсуліну і схильність до розвитку діабету [9]. Передбачається, що накопичення жиру в ПЗ в умовах метаболічного синдрому може призвести до аналогічного процесу, названого неалкогольною жировою хворобою підшлункової залози (НЖХПЗ), із ризиком розвитку стеатопанкреатиту [10].
З огляду на важливість адекватного нутритивного забезпечення та гормонального гомеостазу для динамічного, гармонійного розвитку дитини необхідний кваліфікований підхід до діагностики та лікування захворювань ПЗ у дітей [11].
Термінологія
У літературі існують різні визначення та синоніми для опису акумуляції жиру в ПЗ, вживаються такі терміни, як «стеатоз», «ліпоматоз підшлункової залози», «жирна підшлункова залоза», «жирова інфільтрація», «жирове заміщення ПЗ», «неалкогольна жирова хвороба підшлункової залози», у складі якої виділяють неалкогольний стеатопанкреатит [12].
М. Смітс і співавт. (2011) визнають, що «стеатоз ПЗ» є оптимальним терміном, що характеризує акумуляцію жиру в ПЗ без заміщення тканини ПЗ жировою тканиною, що зумовлює оборотність процесу [10]. Жирове заміщення має принципово інший зміст, що передбачає загибель паренхіматозних клітин і появу замість них жирових (адипоцитів). Зі свого боку, термін «неалкогольна жирова хвороба підшлункової залози» зумовлює зв’язок даного стану з ожирінням, виключаючи вроджену патологію [12].
Надалі в даному огляді ми будемо використовувати поняття «стеатоз підшлункової залози як оборотний процес накопичення жиру в ПЗ» і «неалкогольна жирова хвороба підшлункової залози як компонент метаболічного синдрому».
Історична довідка
Зв’язок між метаболічними порушеннями та жировою інфільтрацією підшлункової залози є доведеним фактом, що був вперше описаний в 1926 році Д. Шеффер, який виявив кореляцію між вагою людського тіла та структурними змінами в підшлунковій залозі [13].
Стеатоз підшлункової залози детально вивчався Р. Огілві, який зазначив дворазове збільшення поширеності стеатозу підшлункової залози (за даними автопсії) у осіб із надлишковою масою тіла [14]. Установлено, що підшлункова залоза в пацієнтів із нормальною масою тіла і в пацієнтів, які страждають від ожиріння, має різний ступінь розвитку жирової тканини: ступінь ліпоматозу був підвищений у пацієнтів з ожирінням (17,1 %) порівняно з контрольною групою (9,3 %). Огілві також виявив гіпертрофію острівців Лангерганса в пацієнтів з ожирінням.
В 1978 році Т. Олсен і співавт. оприлюднили дані дослідження морфологічних змін при стеатозі ПЗ, надали класифікацію ступенів ліпоматозу за вмістом жирової тканини: 1 ступінь ліпоматозу характеризувався одиночними групами жирових клітин у паренхімі підшлункової залози, 4 ступінь представлений практично повною заміною екзокринної тканини жировою тканиною, 2 і 3 ступінь відповідно проявляються проміжним вмістом жирової тканини.
У роботах Т. Олсен і співавт. (1978) описана залежність між вмістом адипоцитів підшлункової залози і віком, а також вагою обстежених [15].
Довгий час стеатоз підшлункової залози розглядався як патологія виключно старшої вікової групи. Однак сьогодні підтверджена можливість розвитку неалкогольної жирової хвороби підшлункової залози в дітей [16].
Етіологія
Більше 90 % людської популяції мають вміст панкреатичної жирової тканини менше 5 % [12]. З огляду на те, що жирова дистрофія є універсальним патологічним процесом, стеатоз ПЗ може спостерігатися при безлічі вроджених і системних захворювань.
Відповідно, можна виділити групи етіологічних факторів стеатозу ПЗ:
1. Метаболічні порушення: ожиріння, метаболічний синдром, білково-енергетична недостатність.
2. Вроджені та спадкові захворювання: муковісцидоз (МВ), синдром Швахмана — Даймонда (SDS), синдром Йохансона — Блізарда (JВS), гемохроматоз, гетерозиготна карбоксилестерліпазна мутація (CEL).
3. Вплив токсичних агентів і лікарських препаратів: глюкокортикоїдів, статинів.
4. Інфекційні агенти: реовірусна інфекція, вірус імунодефіциту людини, хронічний вірусний гепатит.
Екзокринна недостатність ПЗ при ряді генетичних захворювань є наслідком деструкції паренхіми залози з подальшим жировим заміщенням тканини [12].
МВ є автосомно-рецесивним захворюванням, що обумовлене наявністю мутації гена трансмембранного регуляторного білка муковісцидозу (CFTR), який бере участь у виробництві панкреатичного соку. МВ асоційований із згущенням секрету підшлункової залози, що в підсумку призводить до пошкодження тканини підшлункової залози і можливим заміщенням паренхіматозних клітин адипоцитами [16].
SDS є рідкісним вродженим захворюванням, що характеризується екзокринною недостатністю підшлункової залози, порушенням функції кісткового мозку, скелетними аномаліями і низькорослістю. Після муковісцидозу є другою найбільш поширеною причиною екзокринної недостатності підшлункової залози в дітей. При цьому синдромі зазнає мутації ген SBDS, функція якого пов’язана з обміном гуанінових нуклеотидів та збіркою рибосом. Патогенез ушкодження підшлункової залози при даному синдромі залишається неясним [17, 16].
JВS є рідкісним, іноді летальним автосомно-рецесивним вродженим захворюванням, що характеризується порушенням розвитку підшлункової залози, носа та шкіри голови, із затримкою розумового розвитку, втратою слуху. Іноді описується як форма ектодермальної дисплазії. Даний синдром викликається мутаціями в гені UBR1, що кодує один із декількох ферментів убіквітинлігази. Екзокринна недостатність ПЗ при синдромі JBS може додатково бути наслідком вродженої заміни ацинусів жировою тканиною [18].
CEL. Ліпоматоз паренхіми підшлункової залози в носіїв мутації в гені карбоксилестерліпази спостерігається вже на ранніх стадіях діабету, що пояснює наявність екзокринної панкреатичної недостатності при синдромі MODY (maturity-onset diabetes of the young — цукровий діабет дорослого типу в молодих). MODY є рідкісною, сімейною, клінічно і генетично гетерогенною формою діабету, що характеризується початком у молодому віці (як правило, 10–45 років) зі збереженням секреції ендогенного інсуліну, із відсутністю автоімунного ураження В-клітин підшлункової залози, відсутністю ожиріння та резистентності до інсуліну з можливістю екстрапанкреатичних проявів [19, 20].
Вірусні інфекції: вірусні інфекції, зокрема реовірус, можуть призвести до обструкції проток і, отже, некрозу паренхіми з подальшим заміщенням жировою тканиною [12, 16].
Перевантаження залізом: найбільш важливими причинами перевантаження залізом є спадковий гемохроматоз і перевантаження трансфузійним залізом, що може спостерігатись у результаті повторних переливань крові. Перевантаження залізом підшлункової залози призводить до окислювального стресу ацинарних і острівцевих клітин, апоптозу із заміщенням адипоцитами [12, 16].
Ліки. Дані експериментальних досліджень підтверджують, що ліки можуть викликати некроз тканини підшлункової залози і подальше заміщення жировою тканиною, більшість даних отримано на основі експериментальних моделей на тваринах. Описано, що кортикостероїди, гемцитабін, розиглітазон та октреотид можуть бути пов’язані з панкреатичним стеатозом [12, 16].
Хронічні захворювання печінки. М. Сасакі і співавт. припустили, що панкреатичний ліпоматоз може бути викликаний хронічними захворюваннями печінки, які призводять до обструкції протоки ПЗ. Тепер існують повідомлення про окремі випадки стеатозу ПЗ у пацієнтів із хронічним гепатитом В і цирозом печінки, що підтверджують дану гіпотезу [21, 16].
Мальнутриція. Недоїдання, квашіоркор і СНІД пов’язані зі зміною в структурі підшлункової залози, у тому числі з розвитком ліпоматозу підшлункової залози [12, 16].
Панкреатит. Рецидивуючий гострий панкреатит може призвести до зменшення маси паренхіми із заміщенням тканини адипоцитами [16].
Найбільш поширеними причинами стеатозу підшлункової залози в дітей визнаються ожиріння та метаболічний синдром [22]. На сьогодні немає інформації щодо асоціації між відповідним генетичним фоном і схильністю до стеатозу підшлункової залози. Проте генетична схильність, що пов’язана з ожирінням/метаболічним синдромом, може бути залучена до виникнення жирової інфільтрації підшлункової залози [23]. Генетична схильність до ожиріння, діабету й неалкогольної жирової хвороби печінки пов’зана з варіабельністю груп генів:
1) генів, пов’язаних з інсулінорезистентністю (адипонектину, резистину, рецептора інсуліну, рецептора меланокортину-4 (MC4R), γ-рецептора, що активується пероксисомними проліфераторами (PPARy)) [24, 25];
2) генів, пов’язаних з ожирінням (проопіомеланокортину, α- і β-меланоцитстимулюючого гормона (α- і β-MSH), альфа-кетоглутаратзалежної діоксигенази (FTO), нейромедину В (NMB)) [24, 26];
3) генів, відповідальних за печінковий метаболізм вільних жирних кислот (печінкової ліпази, лептину, рецептора лептину, адипонектину мікросомального білка-транспортера тригліцеридів (ММТР), фосфатидилетаноламінметилтрансферази (РЕМТ)) [24];
4) цитокінасоційованих генів (туморнекротизуючого фактора α, інтерлейкіну-10);
5) генів, асоційованих із розвитком й прогресуванням стеатозу печінки (інсулінрегульованої фосфоліпази (PNPLA3), білка-регулятора глюкокінази (GCKR), аполіпопротеїну С3 (АРОСЗ), лізофосфоліпази-1 (LYPLAL1) [24, 27, 28].
Обидві нозології — НАЖХП та НАЖХПЗ асоційовані з ризиком розвитку діабету та підвищенням загального кардіоваскулярного ризику незалежно від віку, статі, ожиріння та інших метаболічних факторів [29], тому визначення генетичного підґрунтя розвитку стеатозу печінки та підшлункової залози є перспективним напрямком.
Епідеміологія
Поширеність НЖХПЗ у загальній популяції наближається до 16 %, за даними популяційного дослідження, проведеного в Гонгконгу [12]. Дослідження, проведене в Джакарті (2015 р.), продемонструвало, що стеатоз підшлункової залози є частою знахідкою серед дорослого населення з частотою виявлення при профілактичному огляді до 35 % [30, 31].
Згідно з даними дослідників із США (2016), стеатоз ПЗ відмічається в 10 % дитячого населення та тісно асоційований з ожирінням і стеатозом печінки [32].
Проте навіть з урахуванням обмежень цих досліджень, а саме орієнтованості на пацієнтів лікарень, усі вони свідчать про високий рівень поширеності cтеатозу ПЗ серед населення загалом.
Даними численних досліджень останніх років підтверджені вірогідні відмінності між ризиком розвитку стеатозу підшлункової залози в різних етнічних групах (іспанський > афроамериканський > кавказький етнос) [33].
Гендерні відмінності розподілу вісцерального жиру та плазмових тригліцеридів були продемонстровані в багатьох дослідженнях. Нільсен і співавт. (2012) припустили, що експорт вільних жирних кислот у кровотік, що є результатом вісцерального ліполізу, активніший у жінок [34, 35].
Однак, за даними Дж. Лі (2009), А. Россі (2011), вміст жирової тканини ПЗ є вищим у представників чоловічої статі незалежно від ІМТ [22, 35, 36].
Таким чином, дані літературних джерел свідчать про значну поширеність стеатозу підшлункової залози, інформація про поширення стеатозу ПЗ у представників різної статі суперечлива і вимагає подальшого вивчення.
Патогенетичні аспекти розвитку ендокринної недостатності підшлункової залози на фоні стеатозу органа
Жирова тканина підшлункової залози розглядається як новий, асоційований з ожирінням фактор, що може сприяти розвитку дисфункції β-клітин. Установлено [37], що вміст панкреатичної жирової фракції позитивно корелює з умістом жирової фракції печінки, а також із рівнем вільних жирних кислот плазми крові (ВЖК).
Альтерація жирової тканини пов’язана зі збільшенням пулу циркулюючих ВЖК, що відіграють важливу роль в ектопічному накопиченні жиру і розвиткові запалення. Клітинний гомеостаз жирних кислот передбачає баланс між механізмами генерації, транспортування та утилізації. У клітинах ссавців вільні жирні кислоти генеруються шляхом синтезу de novo і шляхом гідролізу тригліцеридів і фосфоліпідів клітинними ліпазами. Проте клітини нежирової тканини мають обмежену ємність для зберігання ліпідів. Порушення функцій клітини або її загибель унаслідок надмірної акумуляції ліпідів пояснюється феноменом ліпотоксичності. Ліпідне перевантаження в панкреатичних β-клітинах призводить до дисрегуляції ендокринної функції [36–39]. Поєднана гіперглікемія посилює негативні наслідки підвищеного рівня плазматичних ВЖК на функцію В-клітин; тому більш доречним є термін «глюколіпотоксичність».
У крові жирні кислоти перебувають у етерифікованому (пов’язаному з альбуміном) або неетерифікованому стані, діапазон концентрацій варіює від 100 мкмоль/л до 1 ммоль/л, рівень залежить від часу доби [34]. Хронічне підвищення рівня ВЖК може бути викликано безліччю причин харчового і стресового типу. Порушення метаболізму ВЖК — ключова подія, що призводить до інсулінорезистентності [39, 43]. У проспективному дослідженні М. Маєт і співавт. (1997) виявлено, що порушення толерантності до глюкози асоційоване з високими рівнями ВЖК натщесерце. Автори дійшли висновку, що підвищені рівні ВЖК є предикторами розвитку порушеної толерантності до глюкози незалежно від наявності інсулінорезистентності та порушення секреції глюкози [39].
Надлишок ВЖК може призводити до порушення функції β-клітин різними шляхами:
— через порушення секреції інсуліну;
— активацію запального процесу в β-клітинах та їх апоптоз;
— мітохондріальну дисфункцію та оксидативний стрес;
— стрес ендоплазматичного ретикулуму (ЕПР).
Стеатоз ПЗ та порушення секреції інсуліну
Жирні кислоти та їх похідні діють не тільки як метаболічні субстрати, але головним чином як сигнальні молекули, що впливають на секрецію інсуліну [36]. Гостре збільшення концентрації ЖК призводить до посилення глюкозостимульованої секреції інсуліну. Навпаки, при хронічному впливі високі концентрації ЖК порушують секрецію інсуліну і несприятливо впливають на виживаність β-клітин [37].
Існують переконливі докази зв’язку між ектопічним накопиченням ліпідів і розвитком резистентності до інсуліну. Проте в деяких випадках, таких як «парадокс атлетів», спостерігається відсутність кореляції між ектопічним накопиченням ліпідів і периферичною резистентністю до інсуліну. Дійсно, спортсмени здатні демонструвати високу чутливість до інсуліну при наявності підвищеного рівня внутрішньом’язових жирних кислот [39, 40]. Таким чином, ектопічне накопичення жирних кислот у нежировій тканині може розглядатися як маркер виникнення резистентності до інсуліну, але не як безпосередня причина інсулінорезистентності [41].
Найбільш ранньою реакцією β-клітин на резистентність периферичних тканин до інсуліну є підвищення активності синтезу та секреції інсуліну. Резистентність до інсуліну є станом, при якому існуюча кількість інсуліну викликає субнормальну біологічну реакцію. Зокрема, вона характеризується зниженням здатності інсуліну стимулювати використання глюкози м’язами і недостатнім пригніченням печінкового синтезу і виділення глюкози [45].
Наявність цих змін у препубертаті викликає особливе занепокоєння, тому що резистентність до інсуліну і пов’язані з ним ускладнення можуть посилюватися під впливом статевого дозрівання через фізіологічне зниження чутливості до інсуліну, пов’язане з нормальним перебігом пубертатного періоду [47].
Тривала дія ВЖК може змінювати експресію генів, що кодують ферменти, які відіграють важливу роль у метаболізмі глюкози, зокрема гексокінази. Так, вплив ВЖК збільшує активність гексокінази, що, зі свого боку, здатна зрушити реагування інсуліну на більш низькі концентрації глюкози і викликати підвищену секрецію інсуліну при низькій концентрації глюкози [48]. Є свідчення того, що ВЖК індукують порушення секреції інсуліну шляхом активації рецепторів GPR40. Білок GPR40 (або рецептор вільних жирних кислот 1) у високій концентрації експресується в панкреатичних β-клітинах, а також активується середньо- і довголанцюговими ЖК. Показано, що в людини мутації гена GPR40 із втратою його функції асоціюються з порушенням секреції інсуліну [48, 49].
Виникаюча гіперінсулінемія сприяє збільшенню маси β-клітин в основному за рахунок активації процесу реплікації. Однак на моделях тварин і в людини було продемонстровано, що після маніфестації діабету спостерігається прогресуюче зниження функції і зменшення маси β-клітин. Ключовим регуляторним білком у цих процесах є фактор транскрипції Fox01 (Forkhead). Fox01 належить до сімейства транскрипційних факторів, що контролюють експресію генів, які беруть участь у фундаментальних клітинних процесах, таких як апоптоз, реакція на окислювальний стрес, клітинна проліферація, клітинне диференціювання та регуляція енергетичного метаболізму [40]. Таким чином, здається можливим, що ВЖК можуть інгібувати транскрипцію гена інсуліну і пригнічувати біосинтез інсуліну [42].
Aктивація запального процесу та апоптоз β-клітин
У клітині з жирних кислот, не витрачених на β-окислення, спочатку синтезуються фосфоліпіди, а потім тригліцериди, що акумулюються в цитоплазмі, яка є підґрунтям стеатозу паренхіматозних органів. Тригліцериди можуть бути резервуаром LC-КоА, оскільки β-клітини підшлункової залози експресують гормон-чутливу ліпазу і здійснюють активний ліполіз. LC-КоА накопичуються в цитоплазмі, де вони також можуть бути використані як попередник діацилгліцеролу (ДАГ) або синтезу церамідів [43].
ДАГ є гліцеридом, що складається з двох ланцюгів жирних кислот, ковалентно пов’язаних із молекулою гліцерину. ДАГ, інтермедіат обміну тригліцеридів і фосфоліпідів, є важливим вторинним месенджером у внутрішньоклітинній сигналізації. Різні комбінації із сигнальними молекулами за допомогою ДАГ можуть активувати специфічну протеїнкіназу С (PKC). Активація PKC може підвищити здатність кіназ, таких як JNK і інгібітор кінази кВ, фосфорилювати субстрат 1 рецептора інсуліну (IRS-1) [27], що інгібує дію інсуліну через руйнування взаємодії IRS-1 із рецептором інсуліну [50].
ДАГ є активатором IκB-кінази (IKK), що активізує фактор NF-кВ. IKK є ферментним комплексом, що бере участь у поширенні клітинної відповіді на запалення [41]. IKK може бути активована ВЖК через взаємодію з Toll-подібними рецепторами-4 [51]. Транскрипційний фактор NF-κB є універсальним фактором транскрипції, що контролює експресію генів імунної відповіді, апоптозу і клітинного циклу. Таким чином, активація NF-κB пов’язана з експресією прозапальних генів, розвитком запалення β-клітин, дисфункцією β-клітин та їх апоптозом.
Цераміди можуть бути причиною стресу β-клітин та порушення секреції інсуліну. Експерименти з використанням культури клітин показали, що цераміди можуть пригнічувати поглинання глюкози шляхом блокування транслокації інсулінзалежного білка — переносника глюкози GLUT4 до плазматичної мембрани, що асоційовано зі зниженням чутливості клітин до інсуліну. В осіб з ожирінням, поєднаним з інсулінорезистентністю, рівні цераміду в скелетних м’язах підвищені в 2 рази. Відомо, що церамід також може індукувати апоптоз β-клітин [52, 53].
Надлишок цераміду гальмує передачу інсулінового сигналу за рахунок пригнічення фосфорилювання протеїнкінази B (Ak/PKB). Активація сигналізації Akt/PKB захищає клітини від індукованого вільними жирними кислотами апоптозу і модулює стійкість клітин до стресу ЕПР. Тобто інгібування даного сигнального шляху під впливом церамідів може призводити до апоптозу β-клітин та зниження стійкості до стресу ендоплазматичного ретикулуму [50, 52].
Мітохондріальна дисфункція та оксидативний стрес
Вільні жирні кислоти є джерелом ДАГ та церамідів, також ВЖК можуть бути транспортовані в мітохондрії для окислення до ацетил-КoA. Ацетил-КоА через цикл трикарбонових кислот є джерелом відновлювальних еквівалентів (NADH і FADH2), що віддають електрони для подальшої генерації аденозинтрифосфату в ланцюзі перенесення електронів. Під час перенесення електронів генерується супероксид-іон (О2–), що може викликати окислювальний стрес і потенційну індукцію антиоксидантних елементів реагування для зниження рівня окисного стресу.
Підвищений рівень малонілкоензиму А (обумовлений гіперглікемією) здатний інгібувати активність карнітинпальмітоїлтрансферази-1, що призводить до зменшення мітохондріального β-окислення і подальшого стимулювання внутрішньоклітинного накопичення тригліцеридів [53].
Мітохондріальна дисфункція включає зниження кількості мітохондрій і мітохондріального біогенезу і/або зменшення експресії мітохондріальних окислювальних білків, таких як комплекси ланцюга перенесення електронів. Ці зміни, ймовірно, призводять до зниження окислення субстратів. Зменшений потік електронів через ланцюг перенесення електронів може згодом призвести до витоку електронів і вільних форм кисню з подальшою появою окислювального стресу та пошкодження клітин. На фоні оксидативного стресу може розвиватись мітофагія (видалення пошкоджених мітохондрій, що запобігає загибелі клітин) або апоптоз, що знижує утилізацію ВЖК та призводить до збільшення накопичення ліпідів. Активні проміжні продукти обміну ліпідів, такі як діацилгліцерол і цераміди, здатні викликати пригнічення сигнального шляху інсуліну. Як відомо, β-клітини є дуже чутливими до дії активних форм кисню і реактивних форм азоту. Також окислювальний стрес, як і ДАГ, є активатором IκB-кінази, що активізує фактор NF-кВ, асоційований з експресією прозапальних генів та дисфункцією β-клітин [54].
Стресс ендоплазматичного ретикулуму та дисфункція β-клітин
Слід зазначити, що фізіологічні процеси зсідання поліпептидного ланцюга (фолдинг), дозрівання, зберігання і транспорт білків перебігають в ЕПР. Ендоплазматичний ретикулум є надзвичайно чутливою до змін гомеостазу внутрішньоклітинною структурою, що здійснює контроль якості протеїнів, які проходять тут процес дозрівання (посттрансляційна модифікація і зсідання) перед переходом їх до апарату Гольджі, причому всі білки, що неправильно зсілися, затримуються і обов’язково знищуються. Важливу фізіологічну роль він відіграє в клітинах, пов’язаних із синтезом великої кількості протеїнів для секреції, зокрема в β-клітинах [52]. Відомо, що накопичення неструктурованих білків, граничні відхилення енергетичного балансу (глюко- і ліпотоксичність, гіпоксія та ін.) впливають на просвіт ЕР і можуть індукувати дисфункцію β-клітин. При наявності місфолдингу в ЕПР широко експресуються трансмембранні сигнальні білки: протеїнкіназа РНК (PERK), що активує фактор транскрипції 6 (ATF6) і інозитолзалежний фермент 1 (IRE1) (рис. 2).
/72.jpg)
Ці сигнальні білки активуються шляхом прямого і/або непрямого зв’язування з білками, фолдинг яких порушений. Комбіновані сигнали від IRE1, PERK і ATF6 впливають на регуляцію генів, що кодують шаперони, оксидоредуктазу і цілий ряд ферментів. У результаті активується комплексний сигнальний механізм відповіді неструктурованих білків (unfolded protein response), спрямований на відновлення нормальної функції ЕПР шляхом зменшення активності трансляції, деградації неправильно структурованих білків і посилення транскрипції генів шаперонів (білків, що сприяють фолдингу). Якщо цей механізм не здатний відновити адекватну функцію ЕПР, він запускає клітинний апоптоз β-клітин [55, 56].
У β-клітинах стрес в ЕПР може бути наслідком ліпотоксичності, а також він асоційований із розвитком інсулинорезистентності і запаленням жирової тканини, що спостерігається при ожирінні і ЦД 2 типу. Більше того, ЕПР сам є основним джерелом активних форм кисню, хвиля окисного стресу, що поширюється з ЕПР, також може активувати як шлях JNK/AP-1, так і шлях IKK/NF-кВ, наслідком чого може бути прискорення прогресування резистентності до інсуліну [57].
Ці результати підкреслюють складність взаємозв’язків між стеатозом підшлункової залози та дією інсуліну. Припускається, що взаємозв’язок між стеатозом паренхіматозних органів і поліорганною резистентністю до інсуліну є багатофакторним та неодноспрямованим.
Патогенетичні аспекти екзокринної недостатності підшлункової залози на фоні стеатозу органа
Іншою клінічно значущою стороною стеатозу підшлункової залози є можливість розвитку екзокринної недостатності. Загалом панкреатична екзокринна недостатність підшлункової залози спостерігається приблизно в однієї третини пацієнтів, які страждають від ожиріння [58].
З одного боку, явища мальдигестії і мальабсорбції в даній категорії пацієнтів можуть розглядатися як вторинні. Однак К. Дупонт і співавт. (1989) спостерігали зниження екзокринної функції підшлункової залози в дитячому та підлітковому віці на тлі стеатозу підшлункової залози, пояснюючи це негативним паракринним впливом адипоцитів на функціональний стан ацинарних клітин [59], що особливо сприйнятливі до трофічної та стимулюючої дії інсуліну, через присутність на ацинарних клітинах інсулінових рецепторів, які беруть участь у регуляції синтезу травних ферментів [60]. Як відомо, ацинарні клітини, що розташовані поруч з острівцями (періінсулярні ацинуси), мають більший розмір, ніж інші, містять велику кількість гранул зимогенів, що відомо під назвою «феномен Галло» [60].
У літературних джерелах є докази того, що екзокринна недостатність при стеатозі підшлункової залози може бути асоційована з великим числом фізіологічних і біохімічних процесів, які включають в себе зниження секреції ендогенного інсуліну і його здатності регулювати метаболізм глюкози, зниження експресії мікроРНК амілази, що супроводжується зниженням синтезу та вивільненням цього ферменту, зниження цитозольної концентрації кальцію (Ca2+) і магнію (Mg2+), зниження активності Na+-K+-ATФази і тирозинкінази, а також нечутливість холецистокінінових рецепторів панкреатичних ацинарних клітин [61].
При безпосередньому накопиченні жирових крапель в ацинарних клітинах (що заперечується рядом дослідників) їх дисфункція може пояснюватися ліпотоксичністю [16].
Зі свого боку, ВЖК впливають як на клітини, так і на капіляри ПЗ. У результаті ішемії створюється кисле середовище (ацидоз), при якому посилюється токсичність ВЖК. Автори також вказують на ще один важливий момент — це підвищена в’язкість крові через високий рівень хіломікронів, що може стати причиною порушення мікроциркуляції в ПЗ та її ішемії [60].
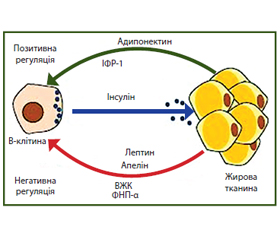
Роль адипоцитокінів у розвитку зовнішньосекреторної та внутрішньосекреторної недостатності підшлункової залози
Відомо, що клітини жирової тканини секретують цитокіни, такі як фактор некрозу пухлини α (ФНП-α), інтерлейкін (ІЛ)-6 і -8, а також виділяють адипокіни, такі як лептин, адипонектин, резистин. Рецептори лептину та адипонектину наявні на β-клітинах ПЗ. Адипоцитокіни є біоактивними пептидами, що модулюють метаболізм інсуліну та катаболізм жиру через внутрішні механізми, відомі як адипоінсулярна вісь (рис. 3). Установлено, що рівень адипонектину при ожирінні знижений, тоді як лептину та резистину — підвищений. Протизапальні ефекти адипонектину включають зниження регуляції прозапальних цитокінів і підвищення регуляції протизапальних цитокінів, зокрема зниження секреції моноцитами ІЛ-6 і ФНП-α. Прозапальна активність лептину проявляється його здатністю вивільняти ІЛ-6 і ФНП-α з макрофагів [58, 61].
/73.jpg)
У дослідженнях in vitro показано, що при стеатозі ПЗ концентрація лептину та інших адипоцитокінів підвищена, особливо в ділянці острівців ПЗ. Паракринний вплив цих молекул не тільки може сприяти дисфункції β-клітин, а також впливає на інкреторну функцію ПЗ [58, 61].
Існують експериментальні докази того, що лептин стимулює окислення жирних кислот, діючи безпосередньо на клітини підшлункової залози, і тим самим обмежує клітинне накопичення тригліцеридів [61, 59].
Патогенетичні аспекти розвитку фіброзу на тлі стеатозу підшлункової залози
Запальні цитокіни, фактори росту (тромбоцитасоційований фактор росту і трансформуючий фактор росту β1), оксидативний стрес, зі свого боку, призводять до активації панкреатичних зірчастих клітин (ПЗК), які відіграють ключову роль у розвитку фіброзу ПЗ. ПЗК розташовані в періацинарному просторі і мають довгі цитоплазматичні відростки, що охоплюють основу ацинуса, становлячи приблизно 3,99 % від загальної кількості клітин підшлункової залози. Вони здатні трансформуватися зі стабільного ліпідовмісного до міофібробластоподібного фенотипу [61, 62].
ПЗК виконують широкий спектр функцій, мають здатність до скорочення, проліферації, можуть синтезувати компоненти позаклітинного матриксу (сприяючи фіброзуванню тканини залози) і впливати на оточуюче клітинне середовище. Активний стан ПЗК характеризується синтезом α-гладком’язового актину, гліального фібрилярного кислого білка, колагену I і III типу, десміну, віментину, металопротеїназ (MMP-1, -2), тканинних інгібіторів металопротеїназ (TIMP-1, -2), а також синтезом протеогліканів і гіалуронової кислоти. Важливо відзначити, що ПЗК самостійно здатні синтезувати цитокіни, такі як ТРФ-β, ІЛ-1, ці спостереження свідчать про існування автокринних циклів, що сприяють збереженню активації ПЗК після припинення дії первинного пошкоджуючого чинника, що пов’язане з розвитком фіброзу органа, втратою функції з подальшим розвитком екзокринної недостатності [62].
Слід зазначити, що ПЗ має великі компенсаторні можливості. Для повного перетравлення жирів досить функціонування 2/3 паренхіми ПЗ, перетравлення білків — 1/2, вуглеводів — 1/10. Тільки при дефіциті протеаз і ліпаз понад 90 % розвиваються клінічні прояви зовнішньосекреторної недостатності. У міру прогресування процесу розвивається функціональна панкреатична недостатність унаслідок втрати функціонуючої паренхіми органа, що виникає в результаті запальної деструкції та фіброзу тканини [63].
Однак у дітей з урахуванням функціональної незрілості залози найбільш часто відмічається відносна недостатність підшлункової залози — це оборотний стан, при цьому структура підшлункової залози не порушена, однак порушується адекватна секреція ферментів.
Висновки
1. Найбільш поширеними причинами стеатозу підшлункової залози є ожиріння та метаболічний синдром, що здатні призводити до формування неалкогольної жирової хвороби підшлункової залози.
2. НЖХПЗ є діагнозом виключення, що потребує ретельного обстеження пацієнта з метою виключення вторинних причин стеатозу.
3. Надлишок вільних жирних кислот асоційований з ектопічним накопиченням жиру з розвитком ліпотоксичності, що проявляється порушенням секреції інсуліну, розвитком мітохондріальної дисфункції, оксидативного стресу, стресу ЕПР, інсулінорезистентності та апоптозу β-клітин, що, зі свого боку, призводить до порушення ендокринної функції підшлункової залози.
4. Порушення екзокринної функції при стеатозі підшлункової залози може пояснюватися ліпотоксичністю та негативним паракринним впливом адипоцитів на ацинарні клітини.
5. Тривалий вплив пошкоджуючих факторів, що секретуються жировою тканиною, асоційований з активацією панкреатичних зірчастих клітин із можливим розвитком фіброзу підшлункової залози.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Report of the commission on ending childhood obesity. Geneva, World Health Organization, 2016 // http:// www.who.int/entity/end-childhood-obesity/publications/echo-report/en
2. Lobstein T. Estimated burden of paediatric obesity and comorbidities in Europe. Part 2. Numbers of children with indicators of obesity-related disease / T. Lobstein, R. Jackson-Leach // International Journal of Pediatric Obesity. — 2006. — № 1. — Р. 33-41.
3. Pizzi M.A. Childhood obesity: effects on children’s participation, mental health, and psychosocial development / M.A. Pizzi, K. Vroman // Occup. Ther. Health Care. — 2013. — № 27. — Р. 99-112.
4. Hedley A.A. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002 / A.A. Hedley, C.L. Ogden, C.L. Johnson et al. // Journal of the American Medical Association. — 2004. — № 291. — Р. 2847-2850.
5. Wang Y. Worldwide trends in childhood overweight and obesity / Y. Wang, T. Lobstein // International Journal of Pediatric Obesity. — 2006. — № 1. — Р. 11-25.
6. Berenson G.S. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study / G.S. Berenson, S.R. Srinivasan, W. Bao et al. // New England Journal of Medicine. — 1998. — № 338. — P. 1650-1656.
7. Chiarelli F. Insulin resistance and obesity in childhood / Francesco Chiarelli, Maria Loredana Marcovecchio // Eur. J. Endocrinol. — № 159. — Р. 67-74. — doi: 10.1530/EJE-08-0245.
8. Miller A.L. Obesity-associated biomarkers and executive function in children / A.L. Miller, H.J. Lee, J.C. Lumeng // Pediatr. Res. — 2015. — № 77. — P. 143-147.
9. Nonalcoholic Steatohepatitis (NASH) Is Associated with a Decline in Pancreatic Beta Cell (β-Cell) Function / S. Mohammad Siddiqui , L. Kai Cheang et al. // Digestive Diseases and Sciences. — 2015. — Vol. 60, Issue 8. — P. 2529-2537.
10. Smits M. The clinical significance of pancreatic steatosis / M.M. Smits, E.J. van Geenen // Nat. Rev. Gastroenterol. Hepatol. — 2011. — № 8. — Р. 169-177.
11. Банадига Н.В. Ферментотерапія в педіатрії: доцільність і виваженість підходів до її проведення / Н.В. Банадига // Здоровье ребенка. — 2014. — № 4(55). — С. 102-107.
12. Prachayakul V. Pancreatic Steatosis: What Should Gastroenterologists Know? / V. Prachayakul, P. Aswakul // J. Pancreas (Online). — 2015, May 20. — № 16(3). — Р. 227-231.
13. Schaefer J.H. The normal weight of the pancreas in the adult human being: A biometric study / J.H. Schaefer // Anat. Rec. — 1926. — Р. 32. — P.119-132.
14. Ogilvie R.F. A quantitative estimation of the pancreatic islet tissue / R.F. Ogilvie // Q.J. Med. — 1937. — Vol. 6. — P. 287-300.
15. Olsen T.S. Lipomatosis of the pancreas in autopsy material and its relation to age and overweight / T.S. Olsen // Acta Pathol. Microbiol. — 1978. — № 86 — Р. 367-373. [PMID: 716899, DOI: 10.1111/ j.1699-0463.1978.tb02058].
16. Exploring the metabolic syndrome: Nonalcoholic fatty pancreas disease / R. Catanzaro, B. Cuffari, A. Italia et al. // World Journal of Gastroenterology. — 2016. — № 22(34). — Р. 7660-7675. — doi:10.3748/wjg.v22.i34.7660.
17. Hassan F. Severe Shwachman-Diamond syndrome and associated CF carrier mutations / F. Hassan, C. Byersdorfer, S. Nasr // Respiratory Medicine CME. — 2010. — № 3(1). — Р. 18-19. — doi:10.1016/j.rmedc.2009.02.001.
18. Johanson-Blizzard syndrome with mild phenotypic features confirmed by UBR1 gene testing / N. Alkhouri, B. Kaplan, M. Kay et al. // World Journal of Gastroenterology : WJG. — № 14(44). — Р. 6863-6866. [http://doi.org/10.3748/wjg.14.6863]
19. Pancreatic Lipomatosis Is a Structural Marker in Nondiabetic Children With Mutations in Carboxyl-Ester Lipase / H. Raeder, I.S. Haldorsen , L. Ersland et al. // Diabetes. — № 56(2). — 2007. —Р. 444-449.
20. Gardner D.S. Clinical features and treatment of maturity onset diabetes of the young (MODY) / D.S. Gardner, E.S. Tai // Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. — № 5. — Р. 101-108. [http://doi.org/10.2147/DMSO.S23353]
21. Sasaki M., Nakanuma Y., Ando H. Lipomatous pseudohypertrophy of the pancreas in a patient with cirrhosis due to chronic hepatitis B // Pathol. Int. — 1998. — 48. — 566-568. [PMID: 9701022, DOI: 10.1111/j.1440-1827.1998.tb03951.x]
22. Pancreatic fat accumulation and its relationship with liver fat content and other fat depots in obese individuals / G. Targher, A.P. Rossi, G.A. Zamboni [et al.] // J. Endocrinol. Invest. — 2012. — № 35. — P. 748-753.
23. Possible Involvement of Pancreatic Fatty Infiltration in Pancreatic Carcinogenesis / Mika Hori, Michihiro Mutoh, Toshio Imai et al. // JOP. J. Pancreas (Online). — 2016, Marсh 07. — № 17(2). — Р. 166-175.
24. Неалкогольна жирова хвороба печінки у дітей (частина I) / Степанов Ю.М., Абатуров О.Є., Завгородня Н.Ю., Скирда І.Ю. та ін. // Гастроентерологія. — 2015. — № 2. — С. 99-107.
25. Association between common polymorphism near the MC4R gene and obesity risk: a systematic review and meta-analysis / B. Xi, G.R. Chandak, Y. Shen et al. // PLoS One. — 2012. — Vol. 7. — P. e45731. [PMID: 23049848]
26. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance / Y.S. Lee, B.G. Challis, D.A. Thompson et al. // Cell Metab. — 2006. — Vol. 3. — P. 135-40. [PMID:16459314]
27. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease / S. Romeo, J. Kozlitina, C. Xing [et al.] // Nat. Genet. — 2008. — Vol. 40. — P.1461-5. [PMID: 18820647]
28. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents / N. Santoro, C. K. Zhang, H. Zhao [et al.] // 2012. — Vol. 55. — PP. 781-9. [PMID: 22105854]
29. The association between nonalcoholic fatty pancreas disease and diabetes / H.Y. Ou, C.Y. Wang, Y.C. Yang et al. // PLoS One. — 2013. — Vol. 8. — P. e62561. [PMID: 23671610]
30. Pancreatic fat accumulation and its relationship with liver fat content and other fat depots in obese individuals / G. Targher, A.P. Rossi, G.A. Zamboni et al. // J. Endocrinol. Invest. — 2012. — № 35. — P. 748-753.
31. Prevalence of Non-Alcoholic Fatty Pancreas Disease (NAFPD) and its risk factors among adult medical check-up patients in a private hospital: a large cross sectional study / C. Lesmana, L.S. Pakasi, S. Inggriani et al. // BMC Gastroenterology. — 2015. — № 15. — P. 174. — doi:10.1186/s12876-015-0404-1.
32. Prevalence of Pancreatic Steatosis at a Pediatric Tertiary Care Center / Pham Y.H., Bingham B.A., Bell C.S. et al. // South Med. J. — 2016. — № 109(3). — P. 196-198.
33. Ethnic Differences in Pancreatic Fat Accumulation and Its Relationship With Other Fat Depots and Inflammatory Markers / Kim-Anne lê, Emily E. Ventura, Jessica q. Fisher et al. // Diabetes Care. — 2011. — Vol. 34. — P. 489.
34. Splanchnic lipolysis in human obesity / S. Nielsen, Z. Guo, C.M. Johnson et al. / J. Clin. Invest. — 2004. — № 113. — Р.1582-1588.
35. Clinical implications of fatty pancreas: Correlations between fatty pancreas and metabolic syndrome / Jun Seok Lee, Sang Heum Kim, Dae Won Jun et al. // World J. Gastroenterol. — 2009. — № 15. — Р. 1869-1875.
36. Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus / Miyazaki Y., Glass L., Triplitt C. et al. // Am. J. Physiol. Endocrinol. Metab. — 2002. — № 283. — Р. 1135-1143.
37. Noninvasive quantification of pancreatic fat in healthy male population using chemical shift magnetic resonance imaging: effect of aging on pancreatic fat content / J. Li, Y. Xie, F. Yuan, B. Song et al. // Pancreas. — 2011. — № 40(2). — Р. 295-9.
38. Biddinger S.B. From mice to men: insights into the insulin resistance syndromes / S.B. Biddinger, C.R. Kahn // Annual Review of Physiology. — 2006. — Vol. 68. — P. 123-158.
39. Вельков В.В. Свободные жирные кислоты — новый маркер инсулинорезистентности и ишемии / В.В. Вельков // Дальневосточный медицинский журнал. — 2008. — № 4. — С. 12-18.
40. Yang Haopeng. The role of fatty acid metabolism and lipotoxicity in pancreatic b-cell injury: Identification of potential therapeutic targets / Haopeng Yang, Xuejun Lin // Acta Pharmaceutica Sinica B. — 2012. — № 2(4). — Р. 396-402.
41. Assimacopoulos-Jeannet F. Fat storage in pancreas and in insulin-sensitive tissues in pathogenesis of type 2 diabetes / F. Assimacopoulos-Jeannet // International Journal of Obesity. — 2004. — № 28. — Р. 53-57.
42. Karpe F. Fatty acids, obesity, and insulin resistance: time for a reevaluation / F. Karpe, J.R. Dickmann, K.N. Frayn // Diabetes. — 2011. — № 60(10). — Р. 2441-2449.
43. Lipid-induced pancreatic — cell dysfunction: focus on in vivo studies / Adria Giacca, Changting Xiao, Andrei I. Oprescu et al. // Am. J. Physiol. Endocrinol. Metab. — 2011. — № 300. — Р. 255-262.
44. Islet β-cell failure in type 2 diabetes — Within the network of toxic lipid / J. Janikiewicz, K. Hanzelka, K. Kozinski // Biochem. Biophys. Res. Commun. — 2015. — Vol. 460(3). — P. 491-496.
45. Papa Feroz R. Endoplasmic Reticulum Stress, Pancreatic b-Cell Degeneration, and Diabetes / Feroz R. Papa // Cold Spring Harb Perspect. Med. — 2012. — № 2. — Р. 1-15.
46. King M.W. Introduction to Insulin Activities / M.W. King // http://themedicalbiochemistrypage.org/insulin-7.php. — 2015.
47. Influence of insulin resistance and body mass index at age 13 on systolic blood pressure, triglycerides, and high-density lipoprotein cholesterol at age 19 / A.R. Sinaiko, J. Steinberger, A. Moran et al. // Hypertension. — 2006. — № 48. — Р. 730-736.
48. Protein Kinase C θ Inhibits Insulin Signaling by Phosphorylating IRS1 at Ser1101J. / Yu Li, Timothy J. Soos, Xinghai Li // Biol. Chem. — 2004. — № 279. — Р. 45304. — doi:10.1074/jbc.C400186200.
49. Hasnain Sumaira Z. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes / Sumaira Z. Hasnain, Johannes B. Prins, Michael A. McGuckin // Mol. Endocrinol. — 2015. — № 56(2). — Р. 33-R54. — doi: 10.1530.
50. Boucher J. Insulin Receptor Signaling in Normal and Insulin-Resistant States / J. Boucher, A. Kleinridders, C.R. Kahn // Cold Spring Harbor Perspectives in Biology. — 2014. — № 6(1). — a009191. [http://doi.org/10.1101/cshperspect.a009191]
51. Chavez Jose A. A Ceramide-Centric View of Insulin Resistance / Jose A. Chavez, Scott A. Summers // Cell. Metabolism. — 2012. — Vol. 15, Is. 5. — P. 585-594.
52. Proinsulin Misfolding and Endoplasmic Reticulum Stress During the Development and Progression of Diabetes / J. Sun, J. Cui, Q. He et al. // Molecular aspects of medicine. — 2015. — Vol. 42. — P. 105-118. — doi:10.1016/j.mam.2015.01.001.
53. Enigmatic ectopic fat: prevalence of nonalcoholic fatty pancreas disease and its associated factors in a Chinese population / C.Y. Wang, H.Y. Ou, M.F. Chen et al. // J. Am. Heart Assoc. — 2014. — № 3(1). — Р. e000297.
54. Montgomery Magdalene K. Mitochondrial dysfunction and ininsulin resistance: an update / Magdalene K and Nigel Turner // Endocr. Connect. — 2015. — № 4. — P. 1-15.
55. Samuel V.T. Lipid-induced insulin resistance: unravelling the mechanism / V.T. Samuel, K.F. Petersen, G.I. Shulman // Lancet. — 2010. — № 375. — P. 2267-2277. — doi: 10.1016/S0140-6736(10)60408-4.
56. Мінченко Д.О. Молекулярні основи розвитку ожиріння та його метаболічних ускладнень у дітей / Д.О. Мінченко // Сучасна педіатрія. — 2015. — № 2(66). — С. 109-112. — doi 10.15574/SP.2015.65.109.
57. Fonseca S.G. Endoplasmic reticulum stress and pancreatic beta cell death / S.G. Fonseca, J. Gromada, F. Urano // Trends in endocrinology and metabolism: TEM. — 2011. — Vol. 22(7). — P. 266-274. — doi:10.1016/j.tem.2011.02.008.
58. Dunmore Simon J. The role of adipokines in β-cell failure of type 2 diabetes / Simon J. Dunmore, James E.P. Brown // Endocrinol. — 216(1). — Р. 37-45. — doi: 10.1530/JOE-12-0278.
59. Стеатоз поджелудочной железы. Подходы к терапии / В.Б. Гриневич, Е.И. Сас, Ю.А. Кравчук, К.В. Матюшенко // Гастроэнтерология С.-П. — 2012. — № 2–3. — С. 5.
60. Самсонова Н.Г. Клинико-функциональное состояние поджелудочной железы при метаболическом синдроме / Н.Г.Самсонова, Л.А. Звенигородская // ЭиКГ. — 2012. — № 11. — С. 96-100.
61. Ткач С.М. Неалкогольная жировая болезнь поджелудочной железы: естественное течение, патогенез, современные подходы к диагностике и лечению / С.М. Ткач // Сучасна гастроентерологія. — 2012. — № 1 (63). — С. 60.
62. Сиренко О.Ю. Панкреатические звездчатые клетки как морфологическая основа развития фиброза поджелудочной железы // Морфологія. — 2010. — ІV, № 1. — С. 5-12.
63. Винокурова Л.В. Функциональная недостаточность поджелудочной железы при хроническом панкреатите: ферментозаместительная терапия, лечебное питание / Л.В. Винокурова, Е.А. Дубцова, Т.В. Попова // Лечащий врач. — 2012. — № 2. — С. 3-9.

/70.jpg)