Гострі респіраторні вірусні інфекції — найбільш поширені захворювання (відомо більше ніж 300 видів респіраторних вірусів), що зустрічаються в будь-якому віці, мають як легкі, так і тяжкі (інколи з інвалідністю та летальністю) клінічні прояви.
Крім вірусу грипу, епідемії можуть спричиняти також аденовіруси, респіраторно-синцитіальний вірус і коронавіруси. Так, коронавірусний близькосхідний респіраторний синдром у 2012 році уразив велику кількість людей на Близькому Сході, у Європі, Північній Африці й Азії [35]. Респіраторно-синцитіальна вірусна (РСВ) інфекція — одна із основних причин пневмонії й бронхіоліту в дітей раннього віку, осіб похилого віку [6] та пацієнтів з імунодефіцитом [23].
Актуальним залишається вивчення механізмів противірусного захисту та розробка напрямів медикаментозної цитопротекції при вірусних інфекціях.
Стрес ендоплазматичного ретикулуму
Ендоплазматичний ретикулум (ЕПР) є принципово важливим місцем для укладки й дозрівання трансмембранних, секреторних і ЕПР-залежних білків. Поліпептидні ланцюжки, синтезовані на поверхні рибосом, прилеглих до гранулярності ЕПР, надходять у його порожнини, де створені унікальні умови для їх обрізання й правильного згортання. В ЕПР лінійні послідовності амінокислот набувають необхідної тривимірної структури, після чого функціонально зрілі протеїни переміщуються в цитозоль [2].
Але процес згортання протеїнів чутливий до будь-яких змін навколишнього мікросередовища. Унаслідок дії ряду шкідливих факторів, у тому числі вірусної інфекції (рис. 1), у просвіті ЕПР накопичуються розгорнуті або неправильно згорнуті білки (unfolded proteins — UPs), що становлять загрозу живій клітині й призводять до стресу ЕПР [37].
Доведено, що стрес ЕПР — типовий молекулярно-патофізіологічний процес, що лежить в основі багатьох, у тому числі інфекційних, захворювань [2, 17, 37]. У клітині у відповідь на стрес ЕПР активується комплекс високоспецифічних внутрішньоклітинних сигнальних шляхів (UPR — unfolded protein response). UPR передає інформацію про стан упаковки білка в просвіті ЕПР до цитоплазми та ядра [14, 27] для відновлення й підтримки гомеостазу в ЕПР.
Білки, які неможливо виправити, відправляються в цитозоль, і там вони зазнають протеасомної деградації ERAD (ER-assisted degradation) [26], тобто відбувається очистка від пошкоджених протеїнів [22].
Адаптивна функція стресу ЕПР
Стрес ЕПР та запуск UPR відіграють важливу роль при вірусних інфекціях, оскільки в інфікованих вірусом клітинах продукується велика кількість вірусних білків [37].
Сучасні дослідження концентруються на аспектах патологічної й фізіологічної ролі UPR.
Адаптивна функція UPR реалізується через основні сигнальні каскади. У стані спокою глюкозорегульований шаперон 78 kDa (glucose-regulated protein GRP 78), інша назва BiP (Binding immunoglobulin protein), пов’язаний з трьома основними сенсорно-сигнальними ензимами в мембрані ЕПР: кіназами PERK (RNA-dependent protein kinase-like ER kinase) й IRE1 (inositol-requiring enzyme 1), а також із фактором транскрипції ATF6 (activating transcription factor 6). Усі ці сенсори мають ЕПР-люмінальний домен, що відчуває присутність незгорнутих або неправильно згорнутих протеїнів, трансмембранний домен і цитозольний функціональний домен (рис. 2). Коли виникає стрес ЕПР, GRP 78 завдяки вищому афінітету до незгорнутих протеїнів відщеплюється й рецептори активуються.
PERK — це трансмембранна серин/треонінова кіназа ЕПР, активний гомодимер якої після відриву від зв’язку з GRP 78 (рис. 2) фосфорилює α-субодиницю фактора, що ініціює трансляцію 2 (translation initiation factor 2 — eIF2α), інактивує його, що забезпечує вимикання загальної трансляції протеїнів, і завдяки цьому відбувається запобігання надмірному згортанню протеїнів [2]. Цей трансляційний контроль є ефективним механізмом зменшення кількості неукладених білків у ЕПР. На підтримку зворотної реакції одночасно за допомогою UPR через Activating transcription factor 4 (ATF4) індукується транскрипція DNA Damage-Inducible Protein 34 (GADD34), a білковий продукт рекрутує протеїнфосфатазу 1 (protein phosphatase 1 — PP1), щоб дефосфорилювати eIF2α-P і усунути ослаблення трансляції. Крім того, PERK забезпечує селективну активацію транскрипції індукованих UPR генів, що кодують шаперони ЕПР, а також антиоксидантні й оксидантдетоксикуючі ензими (наприклад, глутатіон-S-трансферазу) для захисту клітин як від окисного, так і від ЕПР-стресів [2].
ATF6 є регуляторним протеїном, який підвищує експресію індукованих UPR генів. Після звільнення від зв’язку з GRP 78 ATF6 (90 kDa) транспортується в апарат Гольджі, де розщеплюється протеазами S1P і S2P з утворенням активного трансмембранного фактора. Цитозольний фрагмент ATF6 (50 kDa) транслокується до ядра, де активує транскрипцію таргетних генів, які кодують ЕПР-шаперони й компоненти системи ERAD [2, 10, 37].
IRE1 є трансмембранним глікопротеїном ЕПР і має як кіназну, так і рибонуклеазну активність у цитоплазматичному домені. Після дисоціації комплексу з шапероном GRP 78 відбувається активація кінази IRE1 шляхом гомодимеризації й трансавтофосфорилювання. IRE1, виявляючи ендорибонуклеазну активність, активує X-box binding protein 1 (XBP1), який індукує експресію генів, що кодують додатковий синтез шаперонів і полегшують згортання та секрецію протеїнів у ЕПР [2, 10].
Крім того, під впливом XBP1 індукується транскрипція генів, що кодують систему ERAD, це забезпечує деградацію незгорнутих протеїнів. Основну роль у деградації відіграє убіквітин-протеасомна система клітини. Незгорнуті й неправильно згорнуті протеїни ковалентно зв’язуються з молекулою убі–квітину й потім зазнають протеолізу в протеасомному комплексі [26].
Адаптивні шляхи стресу ЕПР функціонують не ізольовано. Відмічається конвергенція ATF6 і PERK, що забезпечує взаємний контроль мішеней обох шляхів [2]. Сигнали, що передаються від ефекторів IRE1, PERK і ATF6, поєднуються в ядрі — відбувається активація транскрипції UPR генів-мішеней.
Усі перераховані вище шляхи роблять внесок у виживання клітин в умовах стресу ЕПР. Їх активація веде до затримки трансляції та деградації неправильно згорнутих білків, експресії молекулярних шаперонів ЕПР, а також розширення мембрани ЕПР для зменшення навантаження білками й збільшення можливостей деградації білків у ЕПР [10].
Стрес ЕПР, окисний стрес, апоптоз
Високоінтенсивний стрес ЕПР і активація UPR можуть призвести до окисного стресу й утворення вільнорадикальних молекул [10]. Тобто окисний стрес можна розглядати як складову частину стресу ЕПР в умовах недостатності вказаних вище механізмів UPR.
Саме з окисним стресом пов’язана цитодеструктивна дія вірусної інфекції. Так, вірус грипу А й активація Toll-like receptors 7 (TLR7) викликають у макрофагах обумовлений NOX2 (NADPH oxidase 2) окисний спалах, що веде до гострого пошкодження легень [32]. Доведено роль FABP5 (Fatty acid-binding protein 5) у патогенезі окисного пошкодження та запалення при вірусі грипу А. Саме дефіцит FABP5 суттєво посилює окисне пошкодження й ліпопер–оксидацію при грипі А, що веде до тканинного запалення [15].
Тривала активація UPR через окисний стрес веде до апоптичної загибелі клітин, при якій IRE1 забезпечує проапоптичну функцію (рис. 2). Крім того, активація UPR індукує експресію CCAAT-enhancer-binding protein homologous protein (CHOP)/GADD153 за допомогою PERK і ATF4 шляхів. CHOP є проапоптичним транскрипційним фактором, який також посилює апоптоз. Крім того, апоптоз запускається й через надмірний вихід у цитоплазму іонів кальцію. Ca2+-сигналізація, у свою чергу, пов’язана із збудженням мембрани, що підвищує чутливість клітин до розпізнавання геномів вірусних частинок, які надійшли в клітину [13].
Так, доведено, що РСВ інфекція викликає генерацію активних форм кисню (АФК), що асоціюється із окисним стресом, пошкодженням легень внаслідок суттєвого зниження експресії антиоксидантних ферментів у дихальних шляхах. Підвищення рівня окисненого глутатіону корелює із тяжкістю хвороби у дітей. У дітей із SpO2 ≤ 92 % виявлено вірогідно вищий рівень окисненого глутатіону й глутатіонпероксидази. Рівень співвідношення глутатіону відновленого/глутатіону окисненого вірогідно корелював із рівнем ІЛ-6, ІЛ-8 та ІЛ-10 [24].
Існуюча протягом тривалого часу точка зору, що АФК лише шкодять біологічним тканинам, була поставлена під сумнів після того, як недавно було виявлено, що АФК можуть служити сигнальними молекулами. Мітогенетичні сигнали, опосередковані утворенням АФК, активують фактори транскрипції, включаючи NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells), антиоксидантні ферменти і Bcl-2. Так, різноспрямовані позаклітинні сигнали від інтерлейкіну-1, TNF-α (Tumor necrosis factor α), H2O2 та інших призводять до розвитку окисного стресу, який супроводжується активацією NF-kB [30].
NF-kB управляє геном антиапоптозу bcl-2 (B-cell lymphoma 2) і проапоптичними факторами bax (as bcl-2-like protein 4) і р53. Ген bcl-2 запобігає загибелі клітини й функціонує як внутрішньоклітинний антиоксидант. Отже, при дії стресових факторів долю клітин визначає баланс між молекулами Bcl-2, Bax Bcl-xL (B-cell lymphoma-extra large)/S. Якщо Вах переважає над Bcl, то він перешкоджає впливу Bcl-2 на апоптоз. Подібним чином надлишок Bcl-xS протидіє Bcl-xL. При цьому p53 активує транскрипцію гена bax через p53-залежні реакції й одночасно знижує рівень експресії bcl-2 [8].
Доведено роль окисного стресу в модуляції відповіді на респіраторну вірусну інфекцію через активацію нуклеарного фактора Nrf2 (Nuclear factor erythroid 2-related factor 2) [30] (рис. 4).
Nrf2 — основний активатор транскрипції антиоксидантних генів через зв’язування з антиоксидантними відповідальними елементами (ARE). У стані спокою Nrf2 прихований у цитоплазмі внаслідок взаємодії з kelch-like ECH-associated protein 1 (Keap1). Keap1 також пов’язаний з E3 убіквітин-протеїн-лігазними комплексом Cul3/Rbx1 (Complex of human cullin 3 (CUL3) with human RING box protein 1 — Rbx1), який убіквітинує Nrf2, що веде до деградації протеасоми [10].
Респіраторна вірусна (наприклад, РСВ) інфекція викликає продукування АФК (ROS) через NOX2, що позитивно регулює сигнальні шляхи через експресію противірусних і протизапальних цитокінів. АФК індукують вивільнення Nrf2 із зв’язку з Keap1. Стабілізується Nrf2 додатковою регуляцією через фосфорилювання й ацетилювання. РСВ інфекція підвищує активність гістондеацетилази, що веде до ядерного деацетилювання Nrf2, викликаючи дисоціацію від антиоксидантних відповідальних елементів і пригнічення антиоксидантних генів [10].
Цитопротекція при окисному стресі
Допомагають клітинам вижити при окисному стресі білки теплового шоку (БТШ), завданням яких є збереження структури інших білків, і автофагія. БТШ зв’язуються з розгорнутим білком і утримують його від занадто швидкого згортання, яке, швидше за все, виявиться неправильним. Якщо ж після декількох спроб білок все одно згортається неправильно, то БТШ направляє його на знищення. В умовах стресу ЕПР, коли ризик порушення структури білків підвищується, роль БТШ посилюється та їх кількість збільшується внаслідок активації ATF6 та IRE1.
Автофагія є найдавнішою системою захисту клітин від вірусної інфекції, від «вторгнення ззовні», оскільки разом із частиною цитоплазми захоплюються віруси [21]. Виділяють два різних типи автофагії — мікро- й макроавтофагію. Перший тип дозволяє направити в лізосому для знищення окремі білкові молекули. Це автофагія, опосередкована шаперонами (chaperone-mediated autophagy). Цей шлях запускається за активної участі одного з БТШ — Hsc70 (70-kDa heat shock cognate protein), який направляє білок, що необхідно знищити, до поверхні лізосоми. Тут знаходиться рецептор LAMP-2A (Lysosomal-associated membrane protein 2), що сприяє деградації білка-мішені в середині лізосоми [21]. Другий тип автофагії пов’язаний з утворенням мембранної структури (автофагосоми) навколо тієї частини клітини, яку передбачається знищити. У цьому процесі відіграють роль білки родини Atg (autophagy related protein). Один з них, LC3 (Microtubule-associated protein 1A/1B-light chain 3), є маркером початку автофагії. Ці білки схожі на убі–квітин [21].
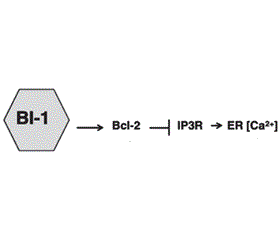
Механізми регуляції виживання клітини в умовах стресу ЕПР подані на рис. 4. BI-1 модулює стрес ЕПР і автофагію через незалежні механізми [28]. BI-1 (Bax inhibitor-1) пригнічує IRE1α, формуючи комплекс з IRE1α, що надалі знешкоджується ендорибонуклеазою (XBP-1) і кіназною активністю (JNK). Шляхом IP3R (Inositol trisphosphate 3 receptor) залежного механізму BI-1 також зменшує стабільний рівень Ca2+ в ЕПР, що спричиняє відповідне зниження рівня мітохондріального Ca2+ й зменшення мітохондріальної біоенергетики. Зменшення рівня АТФ (підвищення АМФ) активує внутрішньоклітинний енергетичний сенсор AMPK (AMP activated protein kinase), який активує автофагію, впливаючи на Atg1 — uncoordinated 51-like kinase 1 (Ulk1)/Ulk2. BI-1 також асоціюється з Bcl-2 у регуляції гомеостазу Ca2+, що також може непрямо впливати на автофагію [28].
Вплив вірусів
на цитопротек–торні захисні механізми
Результати експериментальних досліджень показали, як різні віруси модулюють механізми стресу ЕПР та автофагії, що дозволяє їм уникнути імунної відповіді хазяїна або, що ще гірше, експлуатувати захист хазяїна у своїх інтересах [17].
Встановлено, що зниження активності гістондеацетилази-2 (HDAC2) при окисному й нітрозильному стресі внаслідок респіраторної вірусної інфекції обумовлює загострення хронічного обструктивного захворювання легень [12].
РСВ-інфекція індукує прогресивне зни- ження в ядрі й загалом у клітині рівня транскрипції Nrf2 (через деацетилювання й деградацію Nrf2 протеасомним шляхом), у результаті погіршується зв’язування ендогенних промоторів генів антиоксидантних ферментів і зниження їх експресії [20].
Корекція стресу ЕПР і окисного стресу при респіраторній вірусній інфекції
У наш час вивчаються нові напрями профілактики й терапії респіраторної вірусної інфекції через вплив на механізми стресу ЕПР та окисного стресу. Так, зокрема, перс–пективною є активація шаперону HSP70 (інгібітору каспазної активності й апоптозу). Доведено що експресія HSP70 посилює відповідь на метапневмовірус людини [7].
Інгібітори гістондеацетилази суттєво пригнічують реплікацію РСВ-інфекції та зменшують окисний стрес і запалення дихальних шляхів [11].
Доведено ефективність призначення міметиків антиоксидантів або індукторів Nrf2 при вірус-індукованих хворобах, зокрема при респіраторних інфекціях та інфекціях, асоційованих із зменшенням клітинної антиоксидантної потенції [18]. Інгібітори NOX2 є альтернативою антиоксидантам широкого спектра при лікуванні грипу [34]. Доведено ефективність мелатоніну (за рахунок антиоксидантної дії) при РСВ-інфекції [33].
Найбільш вивчена на даний час антиоксидантна ефективність біофлавоноїдів (рослинних поліфенолів) при лікуванні респіраторної вірусної інфекції. Відомо, що флавоноїди виявляють протизапальну, противірусну, антиоксидантну, антитромбоцитарну, протипухлинну, протиалергічну активність [29].
Проведено доклінічні й клінічні дослідження та доведено противірусні та антиоксидантні властивості препаратів Протефлазід® та Імунофлазід®, що містять флавоноїди диких злаків Herba Deschampsia caespitosa L. та Herba Calamagrostis epigeios L. (НПК «Екофарм», м. Київ, Україна) [1, 3–5]. Методами біохемілюмінесценції та електронно-парамагнітно-резонансної спектроскопії доведено, що флавоноїди даних препаратів по-різному впливають на швидкість генерування супероксидних радикалів нормальними й трансформованими клітинами. При застосуванні їх у низьких дозах (5 мкг/мл) відмічено зниження швидкості генерування супероксидних радикалів клітинами до нуля; високі дози (50 мкг/мл)
у деяких дослідах утримували швидкість генерування супероксидних радикалів клітинами на рівні 0,2 нмоль/105 клітин • хв [4]. Встановлено, що специфічність їх дії зумовлена широким спектром флавоноїдів, які відрізняються ступенем глікозилювання та наявністю різних радикалів у ароматичній частині, що зумовлюють ангіопротекторні, протигіпоксичні ефекти, впливають на дозрівання колагенових волокон, а також виявляють детоксикаційну дію [5]. Профілактичне введення Імунофлазіду гальмувало, а лікувальне — знижувало токсичний ефект вірусу грипу [5].
Добре вивчено антиоксидантні властивості флавоноїду кверцетину [16, 31, 36], який входить до складу вказаних препаратів, у тому числі й через його вплив на обмеження стресу ЕПР [9, 25]. Вважається, що вплив кверцетину на активність антиоксидантної системи опосередковано фактором транскрипції Nrf2 [1].
Лікування з використанням кверцетину при метапневмовірусній інфекції пов’язане із суттєвим зниженням клітинного окисного пошкодження, запальної медіаторної секреції й вірусної реплікації без впливу на транскрипцію вірусних генів і синтезу вірусних протеїнів. Пригнічення вірусної реплікації відбулося на рівні вірусної зборки або вивільнення. Модуляція прозапальної медіаторної експресії відбувається через інгібування NF-κB зв’язування регуляторного фактора 3 інтерферону IRF-3 з його спорідненим сайтом промоторів ендогенних генів [19].
Отже, стрес ендоплазматичного ретикулуму — це накопичення в ньому незгорнутих або неправильно згорнутих білків. Даний патофізіологічний стан виникає в клітині під дією багатьох шкідливих факторів, але при вірусній інфекції набуває особливо важливо значення, оскільки клітина зазнає додаткового навантаження за рахунок синтезу вірусних білків.
Як захисна реакція на стрес ЕПР запускаються процеси:
1) гальмування трансляції білків, щоб запобігти протеїновому навантаженню;
2) селективної активації транскрипції генів, що кодують шаперони ЕПР (білки, що допомагають згортанню білків) та антиоксидантні ферменти;
3) активації деградації «неправильних» білків.
Стрес ЕПР може сприяти або цитопротективній автофагії (процесу самоперетравлювання власних функціонально порушених органел, у тому числі вірусів), або апоптозу (запрограмованій клітинній смерті).
До апоптозу веде окисний стрес (надмірна продукція активних форм кисню), обмовлений тривалим інтенсивним стресом ЕПР і/або недостатністю антиоксидантної системи. Тобто саме окисний стрес є перемикачем від цитопротективної автофагії до апоптозу.
Тому антиоксидантна підтримка патогенетично обґрунтована при лікуванні вірусної інфекції. Біофлавоноїди, підтримуючи антиоксидантні властивості клітини, сприяють цитопротекції в умовах стресу ЕПР та окисного стресу при вірусній інфекції.
В експериментальних дослідженнях in vitro та in vivo доведено антиоксидантні та противірусні властивості препаратів Протефлазід® та Імуно–флазід®, що містять флавоноїди диких злаків Herba Deschampsia caespitosa L. та Herba Calamagrostis epigeios L.
Вказані флавоноїди інгібують також реплікацію вірусів. Отже, пряма противірусна активність у біофлавоноїдів поєднується із антиоксидантними цитопротективними ефектами, що обґрунтовує доцільність їх призначення при вірусній інфекції.
Список литературы
1. Абатуров А.Е., Высочина И.Л. Реализация противовирусного и антиоксидантного действия биофлавоноидов при лечении острых респираторных вирусных инфекций // Здоровье ребенка. — 2016. — 5(73). — 42-48.
2. Зверев Я.Ф., Брюханов В.М. Стресс эндоплазматического ретикулума глазами нефролога (сообщение І) // Нефрология. — 2012. — Т. 16, № 3 (вып. 1). — 54-71.
3. Крамарев С.А. Мета-анализ результатов клинических исследований эффективности флавоноидов при вирусных и вирусно-бактериальных заболеваниях у детей / С.А. Крамарев, А.И. Гриневич, О.Б. Тонковид, О.В. Выговская // Современная педиатрия. — 2014. — № 5. — С. 39-45. — Режим доступу: http://nbuv.gov.ua/UJRN/Sped_2014_5_11.
4. Рыбалко С.Л. Отчет о научно-исследовательской работе «Изучение механизмов действия биологически активных веществ лечебной субстанции Протефлазид». — К., 2010. — 84 с.
5. Сокуренко Л.М. Антитоксична дія імунофлазиду при грипі / Л.М. Сокуренко // Проблеми екологічної та медичної генетики і клінічної імунології. — 2012. — Вип. 6. — С. 408-413.
6. Battles M.B., Langedijk J.P., Furmanova-Hollenstein P., Chaiwatpongsakorn S., Costello H.M., Kwanten L., Vranckx L., Vink P., Jaensch S., Jonckers T.H., Koul A., Arnoult E., Peeples M.E., Roymans D., McLellan J.S. Molecular mechanism of respiratory syncytial virus fusion inhibitors // Nat. Chem. Biol. — 2016 Feb. — 12(2). — 87-93. doi: 10.1038/nchembio.1982. Epub 2015 Dec 7.
7. Baturcam E., Snape N., Yeo T.H., Schagen J., Thomas E., Logan J., Galbraith S., Collinson N., Phipps S., Fantino E., Sly P.D., Spann K.M. Human Metapneumovirus Impairs Apoptosis of Nasal Epithelial Cells in Asthma via HSP70 // J. Innate Immun. — 2016 Oct 11. [Epub ahead of print]
8. Brunelle J.K., Letai A. Control of mitochondrial apoptosis by the Bcl‑2 family // Journal of Cell Science. — 2009. — 122. — 437-441.
9. Cai X., Bao L., Ding Y., Dai X., Zhang Z., Li Y. Quercetin alleviates cell apoptosis and inflammation via the ER stress pathway in vascular endothelial cells cultured in high concentrations of glucosamine // Mol. Med. Rep. — 2016 Dec 15. doi: 10.3892/mmr.2016.6054. [Epub ahead of print]
10. Cervantes-Ortiz S.L., Zamorano Cuervo N., Grandvaux N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology // Viruses. — 2016 May 12. — 8(5). — pii: E124. doi: 10.3390/v8050124.
11. Feng Q., Su Z., Song S., Χu H., Zhang B., Yi L., Tian M., Wang H. Histone deacetylase inhibitors suppress RSV infection and alleviate virus-induced airway inflammation // Int. J. Mol. Med. — 2016 Sep. — 38(3). — 812-22. doi: 10.3892/ijmm.2016.2691. Epub 2016 Jul 26.
12. Footitt J., Mallia P., Durham A.L., Ho W.E., Trujillo-Torralbo M.B., Telcian A.G., Del Rosario A. et al. Oxidative and Nitrosative Stress and Histone Deacetylase‑2 Activity in Exacerbations of COPD // Chest. — 2016 Jan. — 149(1). — 62-73. doi: 10.1378/chest.14-2637. Epub 2016 Jan 6.
13. Hare D.N., Collins S.E., Mukherjee S., Loo Y.M., Gale M. Jr, Janssen L.J., Mossman K.L. Membrane Perturbation-Associated Ca2+ Signaling and Incoming Genome Sensing Are Required for the Host Response to Low-Level Enveloped Virus Particle Entry // J. Virol. — 2015 Dec 30. — 90(6). — 3018-27. doi: 10.1128/JVI.02642-15.
14. Hotamisligil G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease // Cell. — 2010. — 140(6). — 900-917. doi: http://dx.doi.org/10.1016/j.cell.2010.02.034.
15. Gally F., Kosmider B., Weaver M.R., Pate K.M., Hartshorn K.L., Oberley-Deegan R.E. FABP5 deficiency enhances susceptibility to H1N1 influenza A virus-induced lung inflammation // Am. J. Physiol. Lung Cell Mol. Physiol. — 2013 Jul 1. — 305(1). — L64-72. doi: 10.1152/ajplung.00276.2012. Epub 2013 Apr 26.
16. Gomes D.E., Caruso Í.P., de Araujo G.C., de Lourenço I.O., de Melo F.A., Cornélio M.L., Fossey M.A., de Souza F.P. Experimental evidence and molecular modeling of the interaction between hRSV-NS1 and quercetin // Int. J. Biol. Macromol. — 2016 Apr. — 85. — 40-7. doi: 10.1016/j.ijbiomac.2015.12.051. Epub 2015 Dec 21.
17. Jheng J.R., Ho J.Y., Horng J.T. ER stress, autophagy, and RNA viruses // Frontiers in Microbiology. — 2014. — Vol. 5. — 1-13.
18. Komaravelli N., Casola A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses // J. Pharmacogenomics Pharmacoproteomics. — 2014 Sep 30. — 5(4). — pii: 1000141.
19. Komaravelli N., Kelley J.P., Garofalo M.P., Wu H., Casola A., Kolli D. Role of dietary antioxidants in human metapneumovirus infection // Virus Res. — 2015 Mar 16. — 200. — 19-23. doi: 10.1016/j.virusres.2015.01.018. Epub 2015 Jan 30.
20. Komaravelli N., Tian B., Ivanciuc T., Mautemps N., Brasier A.R., Garofalo R.P., Casola A. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2 // Free Radic. Biol. Med. — 2015 Nov. — 88 (Pt B). — 391-403. doi: 10.1016/j.freeradbiomed.2015.05.043. Epub 2015 Jun 11.
21. Kudchodkar S.B., Levine B. Viruses and autophagy // Rev. Med. Virol. — 2009 Nov. — 19(6). — 359-378.
22. McCarthy M.K., Malitz D.H., Molloy C.T., Procario M.C., Greiner K.E., Zhang L., Wang P., Day S.M., Powell S.R., Weinberg J.B. Interferon-dependent immunoproteasome activity du–ring mouse adenovirus type 1 infection // Virology. — 2016 Nov. — 498. — 57-68. doi: 10.1016/j.virol.2016.08.009. Epub 2016 Aug 22.
23. McCutcheon K.M., Jordan R., Mawhorter M.E., Noton S.L., Powers J.G., Fearns R., Cihlar T., Perron M. The Interferon Type I/III Response to Respiratory Syncytial Virus Infection in Airway Epithelial Cells Can Be Attenuated or Amplified by Antiviral Treatment // J. Virol. — 2015 Nov 25. — 90(4). — 1705-17. doi: 10.1128/JVI.02417-15.
24. Moreno-Solís G., de la Torre-Aguilar M.J., Torres-Borrego J., Llorente-Cantarero F.J., Fernández-Gutiérrez F., Gil-Campos M., Túnez-Fiñana I., Pérez-Navero J.L. Oxidative stress and inflamatory plasma biomarkers in respiratory syncytial virus bronchiolitis // Clin. Respir. J. — 2015 Dec 9. doi: 10.1111/crj.12425. [Epub ahead of print]
25. Park E., Chun H.S. Protective effects of quercetin on dieldrin-induced endoplasmic reticulum stress and apoptosis in dopaminergic neuronal cells // Neuroreport. — 2016 Oct 19. — 27(15). — 1140-6. doi: 10.1097/WNR.0000000000000667.
26. Ruggiano A., Foresti O., Carvalho P. ER-associated degradation: Protein quality control and beyond // J. Cell Biol. — 2014. — Vol. 204, № 6. — 869-879.
27. Rutkowski D.T., Hegde R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response // The Journal of Cell Biology. — 2010. — 189(5). — 783-794. doi: http://dx.doi.org/10.1083/jcb.201003138]
28. Sano R., Hou Ying-Chen Claire, Hedvat M., Correa R.G., Shu C. — W. et al. Endoplasmic reticulum protein BI‑1 regulates Ca2+-mediated bioenergetics to promote autophagy // Genes & development. — 2012. — 26. — 1041-1054.
29. Santana F.P., Pinheiro N.M., Mernak M.I., Righetti R.F., Martins M.A., Lago J.H., Lopes F.D., Tibério I.F., Prado C.M. Evidences of Herbal Medicine-Derived Natural Products Effects in Inflammatory Lung Diseases // Mediators Inflamm. — 2016. — 2016. — 2348968. doi: 10.1155/2016/2348968. Epub 2016 Jun 29.
30. Simon P.F., McCorrister S., Hu P., Chong P., Silaghi A., Westmacott G., Coombs K.M., Kobasa D. Highly Pathogenic H5N1 and Novel H7N9 Influenza A Viruses Induce More Profound Proteomic Host Responses than Seasonal and Pandemic H1N1 Strains // J. Proteome Res. — 2015 Nov 6. — 14(11). — 4511-23. doi: 10.1021/acs.jproteome.5b00196. Epub 2015 Oct 9.
31. Teixeira T.S., Caruso Í.P., Lopes B.R., Regasini L.O., Toledo K.A., Fossey M.A., Souza F.P. Biophysical characterization of the interaction between M2-1 protein of hRSV and quercetin // Int. J. Biol. Macromol. — 2017 Feb. — 95. — 63-71. doi: 10.1016/j.ijbiomac.2016.11.033. Epub 2016 Nov 13.
32. To E.E., Broughton B.R., Hendricks K.S., Vlahos R., Selemidis S. Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages // Free Radic. Res. — 2014 Aug. — 48(8). — 940-7. doi: 10.3109/10715762.2014.927579. Epub 2014 Jun 23.
33. Vielma J.R., Bonilla E., Chacín-Bonilla L., Mora M., Medina-Leendertz S., Bravo Y. Effects of melatonin on oxidative stress, and resistance to bacterial, parasitic, and viral infections: a review // Acta Trop. — 2014 Sep. — 137. — 31-8. doi: 10.1016/j.actatropica.2014.04.021. Epub 2014 May 6.
34. Vlahos R., Selemidis S. NADPH oxidases as novel pharmacologic targets against influenza A virus infection // Mol. Pharmacol. — 2014 Dec. — 86(6). — 747-59. doi: 10.1124/mol.114.095216. Epub 2014 Oct 9.
35. Yang Y., Ye F., Zhu N., Wang W., Deng Y., Zhao Z., Tan W. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets // Sci Rep. — 2015 Dec 3. — 5. — 17554. doi: 10.1038/srep17554.
36. Yasui M., Matsushima M., Omura A., Mori K., Ogasawara N., Kodera Y., Shiga M., Ito K., Kojima S., Kawabe T. The Suppressive Effect of Quercetin on Toll-Like Receptor 7-Mediated Activation in Alveolar Macrophages // Pharmacology. — 2015. — 96 (5–6). — 201-9. doi: 10.1159/000438993. Epub 2015 Sep 1.
37. Zhang L., Wang A. Virus-induced ER stress and the unfolded protein response // Front. Plant. Sci. — 2012 Dec 28. — 3. — 293. doi: 10.3389/fpls.2012.00293. eCollection 2012.