Статтю опубліковано на с. 120-128
Вступ
За даними літератури, проблема, що пов’язана як з діагностикою, так і з лікуванням пухлин і пухлиноподібних захворювань скелета у дітей та підлітків, посідає одне з провідних місць поряд з неінфекційними, серцево-судинними захворюваннями та хворобами органів дихання (Всесвітня організація охорони здоров’я (ВООЗ), 2001). Це пов’язано в першу чергу зі значною поширеністю пухлин і пухлиноподібних захворювань в дитячому віці, що становлять 10 % від усіх новоутворень кісток [17].
Окремою нозологічною формою серед пухлин і пухлиноподібних захворювань скелета у дітей виділяється акроформа дисхондроплазії, що пояснюється атиповим перебігом захворювання, швидкою появою та прогресуванням деформацій уражених кісток кистей і стоп, високою частотою малігнізації [40, 63, 65].
Даний огляд літератури присвячено саме цій нозологічній одиниці — акроформі хвороби Ольє і базується на вивченні як історичних аспектів, так і сучасних досягнень медичного сьогодення.
Історія, поширеність, етіологія та патогенез захворювання
Вперше на теренах Європи дисхондроплазію виділив та описав як окреме захворювання відомий французький хірург Луї Ксав’є Едуард Леопольд Ольє (L.X. Ollier) у 1899 році на підставі проведених клініко-рентгенологічних досліджень двох хворих. Він уперше відзначив подібність виявлених ділянок уражених кісток із хрящовою тканиною [4, 6]. Саме йому завдячуємо і терміном «дисхондроплазія», що найбільш вдало відображає дану патологію: «дис» — порушення, «хондро» — хрящ, «плазія» — розвиток, дозрівання.
На вітчизняному просторі перший клініко-рентгенологічний опис хворого чотирнадцяти років з поширеною формою дисхондроплазії належить М.Г. Агаджанову, що було засвідчено протоколами засідання Кавказького медичного товариства 16 жовтня 1897 року, тобто на два роки раніше за L.X. Ollier. Одне з найперших повідомлень про дане захворювання у дорослих належить також нашому співвітчизнику А.А. Рибаковському (1900) [6]. Цим же роком датується дисертація Molin, що був учнем Ollier. У дисертації автор детально розвиває погляди вчителя на сутність дисхондроплазії, проте наводить приклад лише двох спостережень — одного власного та другого, описаного в 1899 році Destot і Nove-Josserand [2].
Сьогодні множинний енхондроматоз, хвороба Ольє (за термінологією ВООЗ (ОМІМ 166000)), або дисхондроплазія, визначається наявністю патологічної хрящової тканини в середині трубчастої кістки та характеризується асиметричним розташуванням хрящових уражень, які можуть бути надзвичайно різноманітними в перерахунку на розмір, кількість, поширення, еволюцію та дебют захворювання [63]. На основі власного матеріалу досліджень (61 хворий з наявністю 961 ураженої кістки) А.А. Аренберг у 1964 р. уперше виділив акроформу як одну з чотирьох основних форм дисхондроплазії, що лягло в основу його класифікації [1].
Поєднання осередків дисхондроплазії, особливо кісток стопи, з ангіоматозними ураженнями шкіри, клітковини і м’язів (множинні ангіоми кавернозного типу) було описане в 1881 році і відоме як синдром Маффуччі (Maffucci syndrome) [5]. Слід мати на увазі, що значно вищий відсоток малігнізації хрящових пухлинних осередків спостерігається при даному синдромі, ніж при хворобі Ольє [2, 15].
Показник поширеності дисхондроплазії становить 1 : 100 000 населення. Оцінка поширеності акроваріанта хвороби Ольє, за даними різних авторів, — від 22,73 [17] до 29,5 % [1].
У ретроспективному плані слід нагадати, що в минулому столітті в медичних колах панували певні погляди щодо ймовірних причин виникнення вищевказаних пухлиноподібних уражень кісток: більшість дослідників пов’язували їх етіологію з порушеннями ембріогенезу в скелеті (Баулина Е.Н., 1975; Virchow R., 1875; Milgram J.W., 1982; Tordai Р. et al., 1990); деякі автори не виключали також ролі травми у виникненні пухлин кісток (Рейнберг С.А., 1964); на думку інших науковців, травма є одним із факторів, що провокують ріст пухлини та сприяють перетворенню ембріональних хрящових ділянок у пухлиноподібну тканину (Волков М.В., 1974).
В.Д. Чаклін (1974) зараховує хворобу Ольє та множинні кістково-хрящові екзостози до групи генотипових хондродисплазій: зазначені порушення хондрогенезу мають деякі загальні риси. В основі обох аномалій лежить «викид» росткової хрящової тканини за нормальні межі зон росту.
На сучасному етапі, з етіологічних позицій, найбільш патогенетично обґрунтованим є визначення дисхондроплазії як патології розвитку скелета, при якій втрачається здатність до нормального енхондрального кісткоутворення, при цьому відбувається утворення хряща пухлинного характеру, з формуванням множинних хондром кісток [17, 43].
Хвороба Ольє не є системним порушенням розвитку в конкретному значенні слова. Зазвичай при суттєвому ураженні одних кісток інші страждають менш значно, а деякі зберігають нормальну будову. Отже, іноді спотворюється диференціювання всього кістково-суглобового апарату (генералізована дисхондроплазія); у деяких пацієнтів спостерігається переважне ураження однієї половини скелета, в інших — залишаються інтактними дистальні відділи скелета, а інколи, навпаки, уражаються головним чином лише кисті та стопи [4].
Встановлено, що енхондральне кісткоутворення порушується при хворобі Ольє переважно вторинно, внаслідок вип’ячування хрящових мас, які розростаються в товщині кістки: іноді в середніх відділах діафіза, в середині кортикального шару, тобто в тій ділянці кісткової тканини, що створюється з окістя, виявляються окремі хрящові включення, не пов’язані з зоною росту. Ймовірно, вони виникають на ранніх етапах ембріогенезу внаслідок патологічного розвитку «надхрящщя», яке ще не перетворилось в окістя; більш імовірна думка щодо їх виникнення з ектопованих хрящових клітин, що виявляються іноді в окісті [9].
Енхондральний тип окостеніння є суворо регульованим процесом, що вимагає прогресії недиференційованих мезенхімальних клітин у гіпертрофовані хондроцити та подальшої заміни хрящової матриці мінералізованою кісткою [42, 60]. Енхондроми розвиваються в метафізарних довгих трубчастих кістках, безпосередньо поблизу росткової пластинки. Відтак було запропоновано (припущено), що вони є результатом порушень сигнальних шляхів контролю проліферації та диференціювання хондроцитів, що призводить до розвитку внутрішньокісткових хрящових осередків.
Генетика
Дані Н.С. Косінської [4] свідчать, що на відміну від множинних кістково-хрящових екзостозів дисхондроплазія, як правило, не успадковується.
Хвороба Ольє та синдром Маффуччі, як вважає К. Сільве (2002), також мають спорадичний характер, не пов’язаний із сімейним типом успадкування.
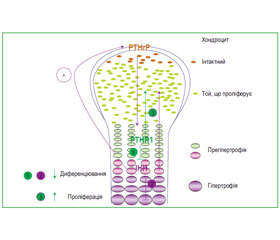
Нерівномірний розподіл уражень при хворобі Ольє наштовхував дослідників на думку, що це — порушення енхондрального формування кісток, яке відбувається через постзиготичні соматичні мутації, що призводять до «мозаїзму»: група науковців-генетиків на чолі з S. Hopyan, а також науковці F. Halal, E.M. Azouz (1991) описують два випадки енхондроматозу синів, у батьків яких були ознаки м’яких скелетних дисплазій, проте без ознак енхондроматозу Ольє. В одному з цих випадків гетерозиготні мутації (R150С) у РТН/РТНrР рецепторів (РТНR1 гена) успадковані від батька [40]. Паратгормон-зв’язаний білок (РТНrР), а також білок — так званий індійський Hedgehog (IHH), що діють на відповідні їм рецептори РТНR1 та PTCH1, надають тісно пов’язані реле сигналізації, що має вирішальне значення в регулюванні енхондральної осифікації (рис. 1).
/122.jpg)
Мутований РТНR1 (R150С) виявився вираженим в енхондромах у двох із шести пацієнтів із множинним енхондроматозом [40]. Мутацію гена виявлено на одній з батьківських алелей в одного пацієнта та його батька, в якого спостерігалась атипова м’яка скелетна дисплазія, проте не хвороба Ольє. Однак ані мутація R150С (26 пухлин), ані жодні мутації РТНR1 гена не можуть бути визначені будь-яким іншим дослідженням, як ті, що призводять до енхондроматозу, зважаючи на неоднорідність молекулярного дефекту [56].
Мутований РТНR1 (R150С), імовірно, конститутивно активізує РТНrР-залежний шлях, таким чином зменшуючи диференціювання хондроцитів, що призводить до формування енхондром [40]. Відповідно до цього висновку, у трансгенних мишей з вираженою мутацією гена РТНR1 під контролем активізатора колагену ІІ типа розвиваються пухлини, що аналогічні тим, які спостерігаються в людському організмі при множинному енхондроматозі. Оскільки при енхондроматозі регулювання IHH за РТНrР виявилось порушеним, були виведені додаткові трансгенні миші, з «надекспресією» Hedgehog (Hh) — транскрипційного регулятора, Gli2. У цих мишей виявлено розвиток ектопічних хрящових острівців, що аналогічні тим, які спостерігалися у мишей з вираженою мутацією РТНR1. Отже, IHH-сигнальний шлях у цілому, напевно, відіграє важливу роль у формуванні енхондром.
Цитогенетика та молекулярна генетика
Існує декілька цитогенетичних доповідей стосовно доброякісного енхондроматозу, проте в них немає жодного посилання на тумор-специфічну хромосому або хромосомну ділянку, що є асоційованою з енхондроматозом або хондросаркомою [24, 25, 57, 58]. Мало відомо про молекулярні механізми, що беруть участь у злоякісній трансформації з енхондроматозу до хондросаркоми. Вираженість (експресія) РТНrР, РТНR1 та їх «підлеглого» партнера Вс12 може бути пов’язаною зі ступенем злоякісності хондросаркоми [48].
Класифікація
У 1964 році А.А. Аренберг створив класифікацію дисхондроплазії, в основі якої лежить принцип переважної анатомічної локалізації осередків ураження [1], що уточнювала та спрощувала попередні класифікації Шінса (1932) та Коччі (1952).
На наш погляд, найзручнішою для розуміння особливостей клінічної картини є класифікація А.П. Крись-Пугача, Р.В. Лучко (1996) за співвідношенням величини та розташування осередків дисхондроплазії:
— великоосередкова центральна;
— великоосередкова крайова;
— дрібноосередкова множинна.
За поширеністю уражень М.В. Волков (1985) розрізняв три форми хвороби Ольє: моноосальну — ураження однієї кістки, олігоосальну — двох-трьох кісток, поліосальну — чотирьох кісток і більше, для якої характерна найтяжча клінічна картина, а також акроформа, при якій уражаються тільки кістки кистей і стоп.
За Міжнародною класифікацією хвороб (ВООЗ, МКХ-10), хвороба Ольє разом із синдромом Маффуччі віднесена до класу «Природжені вади розвитку, деформації кістково-м’язової системи» (шифр Q 87,4.).
Сталий інтерес науковців викликає акроваріант хвороби Ольє з огляду на значний відсоток від загальної кількості хворих на дисхондроплазію, а також відсутність алгоритму хірургічного лікування з урахуванням анатомічної локалізації осередку в ураженій кістці та поширеності останнього, наявності патологічного перелому та осьових деформацій.
Клініка
Класична тріада пухлини вкладається в такі симптоми: біль, порушення функції, збільшення об’єму кістки, що часто тривало відсутні при новоутвореннях у кістках кисті та стопи. На думку Є.Н. Бауліної (1975), можуть спостерігатись як різні варіанти поєднання основних клінічних проявів пухлини, так і повна їх відсутність. Клінічно безсимптомні («німі») пухлини кісток кисті часто виявляються як рентгенографічна знахідка, з приводу травми або захворювань кисті (Веснін А.Г., Семенов І.І., 2002).
Клінічні прояви енхондроматозу Ольє часто з’являються в першій декаді життя і зазвичай розпочинаються з появи веретеноподібних потовщень (іноді з підвищеною чутливістю) на одному або декількох пальцях; кісткових деформацій, що можуть бути пов’язані з патологічними переломами або виникати самостійно [46, 75]. При дисхондроплазії часто уражаються саме короткі трубчасті кістки. Ураження можуть охоплювати декілька кісток, як правило, розподіляючись асиметрично, виключно або переважно торкаючись однієї сторони тіла. Уражені кістки часто вкорочуються та деформуються. Іноді вкорочення кістки може бути єдиною клінічною ознакою захворювання. Зазначені вкорочення кісток часто пов’язані з кутовою деформацією, що може призвести до обмеження обсягу рухів у суміжних суглобах. При фізикальному обстеженні енхондроми виявляються, як правило, на кінцівках, і візуалізуються як вмонтовані маси у фаланги, п’ясткові та плесневі кістки [63].
/123.jpg)
Хоча множинний енхондроматоз був описаний Ольє досить давно, наприкінці XIX століття (відповідно, так утворилась назва «хвороба Ольє»), ще тоді автор підкреслював aсиметричність та випадковість розподілення осередків ураження. Деякі дослідники виділяють два клінічних підтипи дисхондроплазії: енхондроматоз і хворобу Ольє. Першою формою уражаються переважно чоловіки, вона характеризується енхондромами, розташованими в основному на кінцівках та, ймовірно, успадковується за автосомно-домінантним типом [37]. Друга форма уражає переважно жінок, характеризується однобічним розподілом осередків і спостерігається епізодично [63]. За клінічним перебігом дисхондроплазії виділяють 3 основних типи захворювання:
— прогресивний перебіг і виражені клінічні ознаки (саме акроформі властива така клінічна картина);
— непрогресивний перебіг і маловиражені клінічні ознаки;
— «німий» (безсимптомний) перебіг [1].
У хворих з акроваріантом множинного енхондроматозу утворюються щільні бугристі хрящові вузли, що випинаются у м’які тканини, з огляду на відсутність м’язового масиву в кистях і стопах. Пальпаторно — щільні та щільно-еластичні пухлиноподібні утворення, як правило, безболісні; сферичної форми, легко діагностуються. Вони невід’ємно пов’язані з підлеглими ділянками скелета. При значній вираженості цього явища може виникнути певна атрофія шкіри, що вкриває хрящові вузли.
Характерні особливості акроформи дисхондроплазії — це ураження кісток лише кистей і стоп, прогресуючий ріст хрящових вузлів, при якому виявляються веретеноподібне здуття або вузлоподібні потовщення фаланг, п’ясткових і плеснових кісток; іноді осередки патологічної ембріональної тканини пролабують за межі кістки; також характерна швидка поява та наростання тяжких, інвалідизуючих деформацій, що обумовлюють обмеження рухів і порушення функції кистей і стоп, їх спотворення.
Діагностика
Діагностика хвороби Ольє базується на клінічних і традиційних радіологічних дослідженнях.
Клінічний метод обстеження:
— анамнез: початок захворювання і поява скарг, перші симптоми захворювання, залежність їх від травми, виникнення деформацій та обмеження рухів у суміжних суглобах, наявність болю, його інтенсивність і залежність від навантаження;
— ортопедичний статус: поза в спокої, хода (при наявності патологічних осередків у кістках нижніх кінцівок), функція суглоба, наявність вкорочення пальців або кистей чи стоп у цілому, оцінка симетричності м’язової маси;
— функція суглобів: дослідження функції ураженої патологічним процесом кінцівки та уражених суглобів проводиться методом стандартної ангулометрії; обмеження обсягу рухів і контрактура; наявність припухлості; зміна місцевої температури; болючість при пальпації, судинний рисунок у зоні болючості.
Сьогодні основним методом діагностики залишається рентгенологічний. Обсяг рентгенологічного обстеження визначається з огляду на клінічну картину, скарг і допустимих меж опромінення для дітей. У першу чергу обстежуються зони з візуальною наявністю осередків, по-друге, контралатеральні ділянки та інші дистальні сегменти скелета [16, 17].
Множинні енхондроми рідко спостерігаються при народженні, хоча ураження (мікроосередки), ймовірно, є вже наявні. Рентгенологічно зазвичай виявляються декілька осередків ураження у вигляді гомогенних просвітлень овальної або видовженої форми з чітко визначеними, дещо потовщеними кістковими межами [46, 75]. З часом осередки частково звапновуються та стають дифузно-плямистими або пунктирними, можлива легка трабекулярність. Патологічні осередки часто зібрані в скупчення — кластери, в результаті чого метафізарі зони збільшуються. При локалізації патологічної тканини на межі кістки енхондроми створюють типові паз-подібні зображення.
Існують повідомлення щодо незначної затримки кісткового віку дітей (в средньому на 0,6 ± 1,3 року), які страждають від хвороби Ольє [44].
«Осередки Ольє» локалізуються майже винятково в метафізарних зонах довгих трубчастих, а також у коротких кістках рук і ніг. Вони спочатку локалізовані поблизу хрящової зони росту, а потім поступово мігрують у напрямку діафіза; епіфізи, що суміжні з ураженим метафізом, можуть вторинно ушкоджуватись [33, 46].
Важливо підкреслити асиметричність розподілу пухлинних уражень, які можуть бути локалізовані на одній кінцівці або обмежуватися однією половиною тіла. Проте навіть при обмеженні в основному в одній стороні тіла при ретельному дообстеженні одна або дві енхондроми часто виявляються з іншого боку, власне кажучи, на кістках кисті або стопи.
При ураженні кистей і/або стоп майже ніколи не ушкоджуються одразу всі п’ясткові кістки та фаланги. За свідченням А.А. Аренберга, найбільш часто уражаються фаланги пальців кистей (29,3 % від загальної кількості уражених кісток), фаланги пальців стоп (21,0 %), п’ясткові кістки (10,8 %), плесневі кістки (5 %).
Характерними є такі рентгенологічні зміни в кістках при дисхондроплазії: у метафізарних відділах довгих трубчастих кісток виявляються округлі, овальні або видовжені осередки просвітлення кісткової тканини з вираженими межами, спочатку ці осередки локалізуються біля самої межі епіфізарного хряща, в подальшому за умови відсутності зв’язку із зоною росту вони переміщуються в напрямку діафіза (що пов’язано з ростом кістки в довжину), збільшуючись у розмірах, із потенційно можливим руйнуванням кортикального шару кістки. В більш тяжких випадках патологічний процес захоплює росткову зону та епіфізарний хрящ. У коротких трубчастих кістках при ураженні кистей і стоп (акроформа) хрящові осередки займають весь діафіз, викликаючи його веретеноподібне здуття. Часто спостерігаються множинні осередки ураження, що стало причиною виникнення одного з синонімів назви — «множинний енхондроматоз», що частіше зустрічається в англомовних джерелах.
У коротких кістках хрящові маси викликають атрофію внаслідок тиску в кірковій речовині та призводять до деформацій відповідних відділів. На фоні хрящової тканини видно вапняні включення. Розвиток епіфізів та апофізів при дисхондроплазії порушується незначно, однак епіфізи можуть змінюватись вторинно через деформації метафізів [9]. Хрящові маси, що вростають у фаланги, іноді викликають внаслідок тиску повну атрофію кортикального шару, що вкриває їх. Спочатку він розсмоктується в окремих місцях, потім на всьому протязі поверхні вип’яченої ділянки. В поодиноких випадках спостерігається рентгенологічний феномен — трикутник Кодмена, або так званий симптом «козирка», який формується в результаті нависання кортикального шару над хрящовим осередком.
Такі додаткові методи дослідження, як ультразвукове обстеження (УЗО), магнітно-резонансна томографія (МРТ), є надзвичайно інформативними та важливими для оцінки та спостереження динаміки ураження, особливо при появі якісно нових симптомів (таких як збільшення розмірів, виникнення або посилення болю тощо). Необхідно підкреслити, що сонографія дозволяє без додаткового променевого навантаження оцінити структуру м’яких тканин довкола та в середині суглоба, визначити ознаки асептичного запалення в порожнині суглоба. А також отримати додаткові дані про структуру пухлини (щодо диференціальної діагностики), її межі. З огляду на те, що УЗО здійснюється в режимі реального часу, це дає змогу виконувати багатопроекційне дослідження, контролюючи, як змінюється структура зображення залежно від проекції. Внаслідок застосування допплерівського методу дослідження можливо проводити оцінку кровообігу, диференціювання судинних структур, отримувати об’ємне зображення судинного дерева досліджуваного утворення.
Патоморфологічний аналіз має обмежену роль у первинній діагностиці та переважно застосовується при підозрі на малігнізацію процесу. Проте, морфологічне дослідження є обов’язковою складовою комплексного обстеження та кінцевої діагностики даної групи хворих. Всі діагнози у даного контингенту пацієнтів повинні бути верифіковані відповідно до міжнародної гістологічної класифікації [27, 31, 48, 55].
У деяких випадках можливе додаткове застосування імуногістохімічного методу дослідження [73].
Хворобу Ольє необхідно диференціювати від спадкових множинних кістково-хрящових екзостозів [46, 65, 75]. Екзостозна хвороба є автосомно-домінантним захворюванням, характеризується множинним порушенням хондрогенезу та виникає переважно в зонах метафізів довгих кісток. Для встановлення діагнозу зазвичай достатньо клінічного та рентгенологічного досліджень. Найбільш важливим критерієм відмінності енхондроматозу від кістково-хрящових екзостозів є локалізація ураження кістки: екзостози розташовані на поверхні кістки на відміну від осередків енхондроматозу, які розташовані переважно в центрі кістки, що виявляється радіографічно. Диференціальну діагностику також проводять з іншими рідкісними формами хондроматозу, що включають в себе метахондроматоз, спондилоенхондроплазію та генохроматоз І та ІІ типів, що чітко визначені та описані [39, 46].
Гістопатологія
Макроскопічні дослідження осередків зазвичай виявляють кілька округлих або овальної форми хрящових вузликів у кістці [46, 65]. Кожний окремий вузол відмежований по периферії губчастою або пластинчастою кісткою і відокремлений від інших осередків міжтрабекулярним простором. Матрикс хрящових осередків пухлин, як правило, твердий, з міксоїдними змінами, що проявляються (трактуються) як ушкодження матриксу. Для енхондроматозу характерна наявність вражаючої гетерогенності та різноманітності в ступені целюлярності та фенотипу хондроцитів. Таке різномаїття залежить деякою мірою від таких факторів, як локалізація та вік хворого. Певно, через зазначену високу целюлярну гетерогенність визначити різницю гістохімічних крітеріїв між доброякісним множинним енхондроматозом і злоякісною хондросаркомою — досить важко. Гістологічні критерії злоякісності, що застосовуються для виявлення хондросаркоми, не можуть бути застосовані при верифікації хвороби Ольє через високу целюлярність, і, відповідно, визначити різницю між хрящовою пухлиною та хондросаркомою класу А в контексті енхондроматозу надзвичайно важко або навіть неможливо. Тому діагностика базується на поєднанні рентгенівських (деструкція кортикального шару та пролабування в м’які тканини), клінічних і гістологічних критеріїв [46, 63, 65].
Лікування
На ранніх етапах досліджень і спроб лікування уражень кисті та стопи дисхондроплазією деякі учені дотримувалися думки щодо доцільності видалення уражених кісток і навіть виконання ампутації уражених множинними осередками пальців кисті та стопи [1, 13, 15]. Проте, зважаючи на наші спостереження та літературні дані [3, 15, 17], раціональним є лікування шляхом етапного кюретажу хрящеподібної тканини, ретельного заміщення пластичним матеріалом залежно від величини осередка з подальшим моделюванням фаланги при деформації останньої, що спричиняє дисфункцію суміжного суглоба [13].
За В.Д. Чакліном, множинні патологічні осередки в кістках пальців та п’ястка підлягають радикальному вилученню, а іноді проводять діафізарну резекцію кістки на протязі зі збереженням епіфізарних кінців з одномоментним заміщенням дефекту авто- чи гомотрансплантатом.
Максимально ощадне ставлення до суглобового хряща і зон росту та водночас радикальне вилучення пухлинної тканини ставлять не завжди легке завдання перед хірургом. Осередки патологічної хрящової тканини розміром понад 1 см підлягають видаленню, тому що в старшому віці вони можуть бути джерелом розвитку вторинної хондросаркоми [4, 43]. Проте думка нідерландської групи дослідників [63] діаметрально протилежна: вважають оперативне лікування показаним лише за умови появи ускладнень (патологічні переломи, прогресуючий ріст осередка, злоякісна трансформація).
Одним з основних ускладнень енхондроматозу є злоякісна трансформація, отже, необхідно мати настороженість щодо її ознак. Цими ознаками є ерозія кортексу, поширення пухлини в м’які тканини, а також поява «неправильності» або нечіткості поверхні пухлини [63].
M.W. Chapman відзначає, що множинний хондроматоз має мало спільного з солітарними енхондромами, а ураження мають тенденцію до збільшення та пов’язані з деформаціями скелета. Для порівняння: що ризик саркоматозної дегенерації значно більший при дисхондроплазії й має бути запідозреним, якщо пухлини стають болючими, збільшуються або пов’язані з гострою деформацією сегмента [49].
Особливістю перебігу акроформи є найбільша частота малігнізації серед форм дисхондроплазії, що пояснюється відмінністю у гістологічній структурі вогнищ — це найменш диференційовані хрящові вузли [7, 10–62].
Окремим і дискутабельним питанням (для обговорення) є тема показань до застосування пластики щодо утворених пострезекційних дефектів кісток, на якій ми не зупиняємось у цій роботі з огляду на необхідність більш ретельного та поглибленого висвітлення в подальших наших публікаціях.
Прогноз
Прогноз хвороби Ольє важко оцінити [46]. У пацієнта з множинними ураженнями можливий кращий прогноз, ніж у пацієнтів з монофокальним осередком, що може обумовити, наприклад, вкорочення нижньої кінцівки і, таким чином, спричинення асиметрії кінцівок. Особливо, якщо осередок ураження вже є у дуже маленьких дітей. Аналогічно, ранній дебют енхондроматозу при акроформі, зокрема у фалангах, може призводити до виражених деформації пальців. Як зазвичай виявляється, форми з раннім початком є більш агресивними. Таке ускладнення, як компресія суміжних нервів поряд із зонами уражень, зустрічається значно рідше, ніж при множинних кістково-хрящових екзостозах. Щодо патологічних осередків при хворобі Ольє є ризик злоякісної трансформації енхондроми в хондросаркому, яка зазвичай спостерігається в більш ранньому віці, ніж у пацієнтів із солітарною хондросаркомою. Повідомлення про випадки злоякісного перетворення є варіабельними, за різними оцінками, їх частота становить 5–50 % [56, 57, 59, 75]. Ризик малігнізації вищий при синдромі Маффуччі, тому і прогноз тут є серйозніший, ніж при хворобі Ольє [46, 65].
Висновки
Отже, як свідчить огляд ретроспективної та сучасної літератури, у великому розділі дитячої онкоортопедії акроформа — хвороба Ольє внаслідок частоти, тяжкості клінічного перебігу, далеко не завжди легкої та вчасної діагностики, складності лікування, значної частоти різноманітних ускладнень залишається білою плямою. У зв’язку з вищевикладеним актуальність подальшого теоретичного та практичного аспектів дослідження даної патології, безперечно, не викликає сумнівів.
Список литературы
1. Аренберг А.А. Дисхондроплазия костей (клиника, диагностика, лечение): автореф. дис… канд. мед. наук: ЦИТО. — М., 1964. — 16 с.
2. Волков М.В. Болезни костей у детей. — М.: Медицина, 1985.
3. Касымов И.А. Костно-пластические оперативные вмешательства у детей с костной патологией: автореф. дис… д-ра мед. наук. — М., 2000. — 40 с.
4. Косинская Н.С. Нарушения развития косино-суставного апарата. — Л.: Медицина, 1966.
5. Лучко Р.В. Застосування біокомпозитів при оперативному лікуванні акроформи при дисхондроплазії / Р.В. Лучко, А.П. Крисюк // Мат-ли Всеукр. наук.-практ. конференції ортом.-травм. — Київ — Євпаторія, 1998. — С. 129-131.
6. Лучко Р.В. Деформации и укорочения конечностей при дисхондроплазии: дис… канд. мед. наук: УкрНИИТО. — К., 1996. — 200 с.
7. Лучко Р.В. Нове у хірургічному лікуванні акроформи дисхондроплазії // Труды Крымского медицинского университета им. С.И. Геогргиевского. — 1999. — Т. 135, ч. 2.
8. Международная статистическая классификация болезней и проблем, связанных со здоровьем. 10-й пересмотр / Всемирная организация здравоохранения. — М.: Медицина, 1998.
9. Русаков А.В. Патологическая анатомия болезней костной системы. Введение в физиологию и патологию костной системы. — М., 1959.
10. Садыхов А.Г., Мирджавадова А.К., Шизаманов А.М., Злокачественные перерождения некоторых диспластических заболеваний скелета // 5-й Всесоюз. съезд травм.-ортоп. — М., 1988. — Ч. 2. — С. 164-165.
11. Серб С.К. Хирургическое лечение доброкачественных опухолей костей кисти: дис… канд. мед. наук: спец. 14.00.22. Травматология-ортопедия / С.К. Серб. — СПб., 2007. — 141 с.
12. Соловьев Ю.Н. Опухоли костей: Мат-лы к морфологии и патогенезу: автореф. дис… д-ра мед. наук: ИЭиКО АМН СССР. — М., 1970. — 32 с.
13. Усольцева Б.В., Машкара К.И. Хирургия заболеваний и повреждений кисти. — М., 1986. — 3-е изд., перераб. и доп.
14. Утсон Джонс Р. Переломы костей и повреждения суставов: Пер. с англ. — М.: Медицина, 1972. — 672 с.
15. Чаклин В.Д. Опухоли костей и суставов. — М.: Медицина, 1974.
16. Шолохова Н.А. Хирургическое лечение доброкачественных опухолей и опухолеподобных заболеваний внутри- и околосуставной локализации у детей и подростков: автореф. дис… канд. мед. наук: ГОУ ДПО «Российская медицинская академия последипломного образования». — М., 2010. — 19 с.
17. Шолохова Н.А., Моргун В.А., Семенова Л.А. Доброкачественные опухоли и опухолеподобные заболевания костей внутри- и околосуставной локализации у детей (диагностика и лечение) // Детская онкология. — 2007. — № 3–4. — С. 68-75.
18. Alexandre C., Jacinto A., Ingham P.W. Transcriptional activation of hedgehog target genes in Drosophila is mediated directly by the cubitus interruptus protein, a member of the GLI family of zinc finger DNA-binding proteins // Genes. Dev. 1996; 10: 2003-13.
19. Amling M., Neff L., Tanaka S., Inoue D., Kuida K., Weir E., Philbrick W.M., Broadus A.E., Baron R. Bcl-2 lies downstream of parathyroid hormone related peptide in a signalling pathway that regulates chondrocyte maturation during ske–letal development // J. Cell. Biol. 1997; 136: 205-13.
20. Auyeung J., Mohanty K., Tayton K. Maffucci lym-phangioma syndrome: an unusual variant of Ollier’s // J. Pediatr. Orthop. B. 2003; 12: 147-50.
21. Baumgart R., Burklein D., Hinterwimmer S., Thaller P., Mutschler W. The management of leg-length discrepancy in Ollier’s disease with a fully implantable lengthening nail // J. Bone Joint. Surg. Br. 2005; 87: 1000-4.
22. Benbouazza K., El Hassani S., Hassikou H., Guedira N., Hajjaj-Hassouni N. Multiple enchondromatosis: a case report // Joint. Bone Spine. 2002 Mar; 69(2): 236-9.
23. Bertoni F., Bacchini P., Hogendoorn P.C.W. Chondrosarcoma. World Health Organisation classification of tumours / Fletcher C.D.M., Unni K.K., Mertens F., editors // Pathology and genetics of tumours of soft tissue and bone. — Lyon: IARC Press, 2002. — P. 247-51.
24. Bovee J.V., Cleton-Jansen A.M., Kuipers-Dijkshoorn N.J., van den Broek L.J., Taminiau A.H., Cornelisse C.J., Hogendoorn P.C. Loss of heterozygosity and DNA ploidy point to a diverging genetic mechanism in the origin of peripheral and central chondrosarcoma // Genes. Chromosomes Cancer. 1999; 26: 237-246.
25. Bovee J.V., van Roggen J.F., Cleton-Jansen A.M., Taminiau A.H., van der Woude H.J., Hogendoorn P.C. Malignant progression in multiple enchondromatosis (Ollier’s disease): an autopsy-based molecular genetic study // Hum. Patho. 2000; 31: 1299-1303.
26. Bovée J.V.M.G., Van den Broek L.J.CM., Cleton-Jansen A.M., Hogendoorn P.C.W. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma // Lab. Invest. 2000; 80: 1925-33.
27. Bukte Y., Necmioglu S., Nazaroglu H., Kilinc N., Yilmaz F. A case of multiple chondrosarcomas secondary to severe multiple symmetrical enchondromatosis (Ollier’s disease) at an early age // Clin. Radiol. 2005; 60: 1306-10.
28. Couvineau A., Wouters V., Bertrand G., Rouyer C., Gerard B., Boon L.M., Grandchamp B., Vikkula M., Silve C. PTHR1 mutations associated with Ollier disease result in receptor loss of function // Hum. Mol. Genet. 2008; 17: 2766-75.
29. D’Angelo L., Massimi L., Narducci A., Di R.C. Ollier disease // Childs Nerv. Syst. 2009; 25: 647-53.
30. Eefting D., Schrage Y.M., Geirnaerdt M.J., Le Cessie S., Taminiau A.H., Bovee J.V.M.G., Hogendoorn P.C.W. Assessment of interobserver variability and histologic parameters to improve reliability in classification and grading of central cartilaginous tumors // Am. J. Surg. Pathol. 2009; 33: 50-7.
31. Flemming D.J., Murphey M.D. Enchondroma and chondrosarcoma // Semin. Musculoskelet. Radiol. 2000; 4: 59-71.
32. Herget G.W., Strohm P., Rottenburger C., Kontny U., Krauß T., Bohm J., Sudkamp N., Uhl M. Insights into Enchondroma, Enchondromatosis and the risk of secondary Chondrosarcoma. Review of the literature with an emphasis on the clinical behaviour, radiology, malignant transformation and the follow up // Neoplasma. 2014; 61, 4: 365-378.
33. Gabos P.G., Bowen J.R. Epiphyseal-metaphyseal enchondromatosis. A new clinical entity // J. Bone Joint. Surg. Am. 1998; 80: 782-792.
34. Geirnaerdt M.J., Hogendoorn P.C.W., Bloem J.L., Taminiau A.H.M., Van der Woude H.J. Cartilaginous tumors: fast contrastenhanced MR imaging // Radiology. 2000; 214: 539-46.
35. Goto T., Motoi T., Komiya K., Motoi N., Okuma T., Okazaki H., Takatori Y., Tange T., Nakamura K. Chondrosarcoma of the hand secondary to multiple enchondromatosis; report of two cases // Arch. Orthop. Trauma. Surg. 2003 Feb; 123(1): 42-7.
36. Haga N., Nakamura K., Taniguchi K., Nakamura S. Enchondromatosis with features of dysspondyloenchondromatosis and Maffucci syndrome // Clin. Dysmorphol. 1998; 7: 65-8.
37. Halal F., Azouz E.M. Generalized enchondromatosis in a boy with only platyspondyly in the father // Am. J. Med. Genet. 1991; 38: 588-592.
38. Halal F., Azouz E.M. Generalized enchondromatosis in a boy with only platyspondyly in the father // Am. J. Med. Genet. 1991; 38: 588-92.
39. Herman T.E., Chines A., McAlister W.H., Gottesman G.S., Eddy M.C., Whyte M.R. Metachondromatosis: Report of a family with facial features mildly resembling trichorhinophalangeal syndrome // Pediatr. Radiol. 1997; 27: 436-441.
40. Hopyan S., Gokgoz N., Poon R., Gensure R.C., Yu C., Cole W.G., Bell R.S., Juppner H., Andrulis I.L., Wunder J.S., Alman B.A. A mutant PTH/PTHrP type I receptor in enchondromatosis // Nat. Genet. 2002; 30: 306-310.
41. Kozlowski K.S., Masel J. Distinctive enchondromatosis with spine abnormality, regressive lesions, short stature, and coxa vara: importance of long-term follow-up // Am. J. Med. Genet. 2002; 107: 227-32.
42. Kronenberg H.M. Developmental regulation of the growth plate // Nature. 2003; 423: 332-336.
43. Liu J., Hudkins P., Swee R. at al. Bone sarcomas associated with Ollier’s deseasse // Cancer. 1987; 59: 1376-1385.
44. Loder R.T., Sundberg S., Gabriel K., Mehbod A., Meyer C. Determination of bone age in children with cartilaginous dysplasia (multiple hereditary osteochondromatosis and Ollier’s enchondromatosis) // J. Pediatr. Orthop. 2004; 24: 102-108.
45. Lucas D.R., Bridge J.A. Chondromas: enchondroma, periosteal chondroma,and enchondromatosis. World Health Organization classification of tumours / Fletcher C.D.M., Unni K.K., Mertens F., editors // Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002, pp. 237-40.
46. Maroteaux P., Le Merrer M. Les maladies osseuses de l’enfant. Paris: Medecine-Sciences, Flammarion; 2002.
47. Märtson A., Haviko T., Kirjanen K. Extensive limb lengthening in Ollier`s disease: 25-year follow-up Medicina (Kaunas). 2005; 41(10): 861-6.
48. Mertens F., Unni K.K. Enchondromatosis: Ollier disease and Maffucci syndrome. World Health Organization Classification of Tumours / Fletcher C.D.M., Unni K.K., Mertens F., editors // Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002, pp. 356-7.
49. Michael W. Chapman, MD Operative Orthopaedics, Second Edition, Volume 2, Philadelphia.
50. Pandey R., White S.H., Kenwright J. Callus dis traction in Ollier’s disease // Acta Orthop. Scand. 1995; 66: 479-80
51. Pannier S., Legeai-Mallet L. Hereditary multiple exostoses and enchondromatosis. Best Practice & Research Clinical Rheumatology. 2008 Mar; 22(1): 45-54.
52. Pansuriya T.C., Bovée J.V.M.G. Enchondromatosis // Atlas Genet Cytogenet. Oncol. Haematol. July 2008.
53. Ranger A., Szymczak A. Do intracranial neoplasms differ in Ollier disease and maffucci syndrome? An in-depth analysis of the literature // Neurosurgery. 2009 Dec; 65(6): 1106-13; discussion 1113-5.
54. Rozeman L.B., Hameetman L., van Wezel T., Taminiau A.H.M., Cleton-Jansen A.M., Hogendoorn P.C.W., Bovée J.V.M.G. cDNA expression profiling of central chondrosarcomas: Ollier disease resembles solitary tumors and alteration in genes coding for energy metabolism with increasing grade. J. Pathol. 2005; 207: 61-71.
55. Rozeman L.B., Hogendoorn P.C.W., Bovée J.V.M.G. Diagnosis and prognosis of chondrosarcoma of bone // Expert. Rev. Mol. Diagn. 2002; 2: 461-72.
56. Rozeman L.B., Sangiorgi L., Briaire-de Bruijn I.H., Mainil-Varlet P., Bertoni F., Cleton-Jansen A.M., Hogendoorn P.C., Bovée J.V. Enchondromatosis (Ollier disease, Maffucci syndrome) is not caused by the PTHR1 mutation p.R150C // Hum. Mutat. 2004; 24: 466-473.
57. Sandberg A.A., Bridge J.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: chondrosarcoma and other cartilaginous neoplasms // Cancer. Genet. Cytogenet. 2003; 143: 1-31.
58. Sandberg A.A. Genetics of chondrosarcoma and related tumors // Curr. Opin. Oncol. 2004; 16: 342-354.
59. Schaison F., Anract P., Coste F., De Pinieux G., Forest M., Tomeno B. Chondrosarcoma secondary to multiple cartilage diseases. Study of 29 clinical cases and review of the literature // Rev. Chir. Orthop. Reparatrice Appar. Mot. 1999; 85 :834-845.
60. Schipani E., Provot S. PTHrP, PTH, and the PTH/PTHrP receptor in endochondral bone development // Birth. Defects Res. Part. C. Embryo Today. 2003; 69: 352-362.
61. Schrage Y.M., Hameetman L., Szuhai K., Cleton-Jansen A.M., Taminiau A.H.M., Hogendoorn P.C.W., Bovée J.V.M.G. Aberrant heparan sulfate proteoglycan localization, despite normal exostosin, in central chondrosarcoma // Am. J. Pathol. 2009; 174: 979-88.
62. Schwartz H.S., Zimmerman N.B., Simon M.A., Wroble R.R., Millar E.A., Bonfiglio M. The malignant potential of enchondromatosis // J. Bone Joint. Surg. Am. 1987; 69: 269-274.
63. Silve C., Juppner H. Ollier disease // Orphanet. J. Rare Dis. 2006; 1: 37.
64. Spranger J.W., Brill P.W., Poznanski A.K. Bone Dysplasias, An Atlas of Genetic Disorders of Skeletal Develop–ment; second ed. New York: Oxford University Press; 2002, pp. 554-70.
65. Twinkal C. Pansuriya, Herman M. Kroon, Judith V.M.G. Bovée J.V.M.G. Enchondromatosis: insights on the different subtypes // Int. J. Clin. Exp. Pathol. 2010; 3(6): 557-569.
66. Unni K.K. Cartilaginous lesions of bone // J. Orthop. Sci. 2001; 6: 457-472.
67. Urist M.R. A 37-year follow-up evaluation of multiplestage femur and tibia lengthening in dyschondroplasia (enchondromatosis) with a net gain of 23.3 centimeters // Clin. Orthop. Relat. Res. 1989: 1; 37-57.
68. Van L.P., Lammens J. Malformation of the humerus in a patient with Ollier disease treated with the Ilizarov technique // J. Shoulder. Elbow. Surg. 2008; 17: e9-11.
69. Vázquez-García B., Valverde M., San-Julián M. Ollier disease: benign tumours with risk of malignant transformation. A review of 17 cases // An Pediatr (Barc). 2011 Mar; 74(3): 168-73.
70. Verdegaal S.H.1, Bovée J.V., Pansuriya T.C., Grimer R.J., Ozger H., Jutte P.C., San Julian M., Biau D.J., van der Geest I.C., Leithner A., Streitbürger A., Klenke F.M., Gouin F.G., Campanacci D.A., Marec-Berard P., Hogendoorn P.C., Brand R., Taminiau A.H. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: an international multicenter study of 161 patients // Oncologist. 2011; 16(12).
71. Veth R., Schreuder B., van Beem H., Pruszczynski M., de Rooy J. Cryosurgery in aggressive, benign, and low-grade malignant bone tumours // Lancet Oncol. 2005; 6: 25-34.
72. Vortkamp A., Lee K., Lanske B., Segre G.V., Kronenberg H.M., Tabin C.J. Regulation of rate of cartilage differentiation by indian hedgehog and PTH-related protein // Science. 1996; 273: 613-22.
73. Walid M.S., Troup E.C. Cerebellar anaplastic astrocytoma in a teenager with Ollier Disease // J. Neurooncol. 2008; 89: 59-62.
74. Watanabe K., Tsuchiya H., Sakurakichi K., Yamashiro T., Matsubara H., Tomita K. Treatment of lower limb deformities and limb-length discrepancies with the external fixator in Ollier’s disease // J. Orthop. Sci. 2007 Sep; 12(5): 471-5.
75. Whyte M. Acquired Disorders of Cartilage and Bone. Washington DC: American Society for Bone and Mineral Research; 2003.