
Журнал «Здоровье ребенка» 1 (69) 2016
Вернуться к номеру
Транзиторный неонатальный сахарный диабет, ассоциированный с нарушением импринтинга хромосомы 6q24. Часть 2. Эпидемиология, этиология и патогенез
Авторы: Абатуров А.Е., Кривуша Е.Л. - ГУ «Днепропетровская медицинская академия Министерства здравоохранения Украины»; Ивашина В.И., Логвинов Д.В., Турова С.В. - КУ «Днепропетровская ГДКБ № 1» ДОС
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
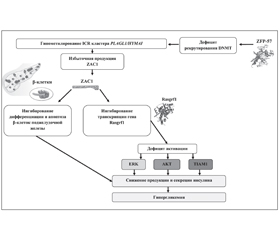
В статье описаны генетические варианты транзиторного неонатального сахарного диабета, указана частота встречаемости различных генетических дефектов у больных с синдромом 6q24-TNDM. Подробно представлены механизмы генетических нарушений, приводящих к развитию данного заболевания. Показано, что развитие 6q24-TNDM ассоциировано с отцовской однородительской дисомией по хромосоме 6, несбалансированной дупликацией q24 на копии отцовской хромосомы 6 и гипометилированием ICR на копии материнской хромосомы 6q24. Дефицит экспрессии гена фактора транскрипции PDX-1 играет важную роль в регенерации поджелудочной железы и дифференцировке β-клеток, а высокая скорость пролиферации и апоптоза клеток сопровождается нестабильностью равновесия продукции инсулина и потребности в нем. Указана роль протеина ZAC1 в патогенезе заболевания.
У статті описані генетичні варіанти транзиторного неонатального цукрового діабету, вказана частота зустрічаємості різних генетичних дефектів у хворих із синдромом 6q24-TNDM. Детально представлені механізми генетичних порушень, що призводять до розвитку даного захворювання. Показано, що розвиток 6q24-TNDM асоційований з однобатьківською дисомією по хромосомі 6, незбалансованою дуплікацією q24 на копії батьківської хромосоми 6 та гіпометилюванням ICR на копії материнської хромосоми 6q24. Дефіцит експресії гена фактора транскрипції PDX-1 відіграє важливу роль у регенерації підшлункової залози та диференціюванні β-клітин, а висока швидкість проліферації й апоптозу клітин супроводжується нестабільністю рівноваги продукції інсуліну та потреби в ньому. Вказана роль протеїну ZAC1 в патогенезі захворювання.
This article describes the genetic variants of transient neonatal diabetes, the incidence of different genetic defects in patients with 6q24-TNDM syndrome. There were presented in detail the mechanisms of genetic disorders leading to this disease. It is shown that the development of 6q24-TNDM is associated with paternal uniparental disomy of chromosome 6, unbalanced q24 duplication on the copy of paternal chromosome 6 and ICR hypomethylation on the maternal copy of chromosome 6q24. Deficiency of gene expression of transcription factor PDX-1 plays an important role in the regeneration of the pancreas and in the differentiation of β-cells, and high rate of proliferation and apoptosis of the cells is associated with imbalance in the production of insulin and the need for it. There was indicated the role of ZAC1 protein in the pathogenesis of the disease.
транзиторный неонатальный сахарный диабет, инсулин.
транзиторний неонатальний цукровий діабет, інсулін.
transient neonatal diabetes, insulin.
Статья опубликована на с. 126-132
Определение синдрома
Эпидемиология
Этиология
/127.jpg)
/128_2.jpg)
Патогенез
/128.jpg)
/130.jpg)
1. Temple I.K., Shield J.P. 6q24 transient neonatal diabetes // Rev. Endocr. Metab. Disord. 2010 Sep; 11(3): 199-204. doi: 10.1007/s11154-010-9150-4.
2. Rubio-Cabezas O., Ellard S. Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options // Horm Res. Paediatr. 2013; 80(3): 137-46. doi: 10.1159/000354219
3. Кураева Т.Л., Емельянов А.О. Клиническая и генетическая гетерогенность неонатального сахарного диабета // Сахарный диабет. 2009; 3: 10-15.
4. Shield J.P. Neonatal diabetes: how research unravelling the genetic puzzle has both widened our understanding of pancreatic deve–lopment whilst improving children's quality of life // Horm Res. 2007; 67(2): 77-83. doi: 10.1159/000096354
5. Hutchison J.H., Keay A.J., Kerr M.M. Congenital temporary diabetes mellitus // Br. Med. J. 1962 Aug 18; 2(5302): 436-40. PMID: 14450256
6. Stanik J. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers / J. Stanik, D. Gasperikova, M. Paskova et al. // J. Clin. Endocrinol. Metab. 2007 Apr; 92(4): 1276-82. doi: http: //dx.doi.org/10.1210/jc.2006-2490.
7. Wiedemann B. Incidence of neonatal diabetes in Austria-calculation based on the Austrian Diabetes Register / B. Wiedemann, E. Schober, T. Waldhoer et al. // Pediatr. Diabetes. 2010 Feb; 11(1): 18-23. doi: 10.1111/j.1399-5448.2009.00530.x.
8. Polak M., Shield J. Neonatal and very-early-onset diabetes mellitus // Semin Neonatol. 2004 Feb; 9(1): 59-65. doi: http: //dx.doi.org/10.1016/S1084-2756(03)00064-2.
9. Flanagan S.E. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood / S.E. Flanagan, A.M. Patch, D.J. Mackay et al. // Diabetes. 2007 Jul; 56(7): 1930-7. doi: 10.2337/db07-0043
10. Aguilar-Bryan L., Bryan J. Neonatal diabetes mellitus // Endocr Rev. 2008 May; 29(3): 265-91. doi: 10.1210/er.2007-0029.
11. Slingerland A.S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births / A.S. Slingerland, B.M. Shields, S.E. Flanagan et al. // Diabetologia. 2009 Aug; 52(8): 1683-5. doi: 10.1007/s00125-009-1416-6.
12. Iafusco D. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1: 90,000 live births / D. Iafusco, O. Massa, B. Pasquino et al. // Acta Diabetol. 2012 Oct; 49(5): 405-8. doi: 10.1007/s00592-011-0331-8.
13. Milenkovic T. Transient neonatal diabetes mellitus in an infant with paternal uniparental disomy of chromosome 6 including he–terodisomy for 6q24 / T. Milenkovic, J. Martic, D.O. Robinson et al. // J. Pediatr. Endocrinol. Metab. 2006 Nov; 19(11): 1353-7. PMID: 17220064.
14. Prando C. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-gamma receptor 1 deficiency / C. Prando, S. Boisson-Dupuis, A.V. Grant et al. // Am. J. Med. Genet. A. 2010 Mar; 152A(3): 622-9. doi: 10.1002/ajmg.a.33291.
15. Suzuki S. Partial paternal uniparental disomy of chromosome 6 in monozygotic twins with transient neonatal diabetes mellitus and macroglossia / S. Suzuki, D. Fujisawa, K. Hashimoto et al. // Clin. Genet. 2010 Dec; 78(6): 580-4. doi: 10.1111/j.1399-0004.2010.01433.x.
16. Temple I.K., Shield J.P. Transient neonatal diabetes, a disorder of imprinting // J. Med. Genet. 2002 Dec; 39(12): 872-5. doi: 10.1136/jmg.39.12.872.
17. Court F. Genome-wide allelic methylation analysis reveals di–sease-specific susceptibility to multiple methylation defects in imprin–ting syndromes / F. Court, A. Martin-Trujillo, V. Romanelli et al. // Hum. Mutat. 2013 Apr; 34(4): 595-602. doi: 10.1002/humu.22276.
18. Strogantsev R. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression / Strogantsev R., Krueger F., Yamazawa K. et al. // Genome Biol. 2015 May 30; 16(1): 112. doi: 10.1186/s13059-015-0672-7.
19. Temple I.K., Mackay D.J.G., Docherty L.E. Diabetes Mellitus, 6q24-Related Transient Neonatal / Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K. // GeneReviews® [Internet]. — Seattle (WA): University of Washington, Seattle; 1993–2015. 2005 Oct 10 [updated 2015 Jan 15]. PMID: 20301706.
20. Mitchell B.D., Pollin T.I. Genomic imprinting in diabetes // Genome Med. 2010 Aug 23; 2(8): 55. doi: 10.1186/gm176.
21. Mackay D.J. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus / D.J. Mackay, S.E. Boonen, J. Clayton-Smith et al. // Hum. Genet. 2006 Sep; 120(2): 262-9. doi: 10.1007/s00439-006-0205-2.
22. Rezwan F.I. A statistical method for single sample analysis of HumanMethylation450 array data: genome-wide methylation analysis of patients with imprinting disorders / F.I. Rezwan, L.E. Docherty, R.L. Poole et al. // Clin. Epigenetics. 2015 Apr 21; 7(1): 48. doi: 10.1186/s13148-015-0081-5.
23. Mackay D.J. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57 / D.J. Mackay, J.L. Callaway, S.M. Marks et al. // Nat Genet. 2008 Aug; 40(8): 949-51. doi: 10.1038/ng.187.
24. Boonen S.E. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings / S.E. Boonen, S. Pörksen, D.J. Mackay et al. // Eur. J. Hum. Genet. 2008 Apr; 16(4): 453-61. doi: 10.1038/sj.ejhg.5201993.
25. Rodríguez-Henche N. Transcription of the mouse PAC1 receptor gene: cell-specific expression and regulation by Zac1 / N. Rodríguez-Henche, F. Jamen, C. Leroy, J. Bockaert, P. Brabet // Biochim. Biophys. Acta. 2002 Jun 7; 1576(1-2): 157-62. doi: 10.1016/S0167-4781(02)00303-2.
26. Mackay D.J. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus / D.J. Mackay, A.M. Coupe, J.P. Shield et al. // Hum. Genet. 2002 Feb; 110(2): 139-44. doi: 10.1007/s00439-001-0671-5.
27. Ma D. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM / D. Ma, J.P. Shield, W. Dean et al. // J. Clin. Invest. 2004 Aug; 114(3): 339-48. doi: 10.1172/JCI200419876.
28. Hoffmann A., Spengler D. Transient neonatal diabetes mellitus gene Zac1 impairs insulin secretion in mice through Rasgrf1 // Mol. Cell. Biol. 2012 Jul; 32(13): 2549-60. doi: 10.1128/MCB.06637-11.
29. Fernández-Medarde A., Santos E. The RasGrf family of mammalian guanine nucleotide exchange factors // Biochim. Biophys. Acta. 2011 Apr; 1815(2): 170-88. doi: 10.1016/j.bbcan.2010.11.001.
30. Veluthakal R. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells / R. Veluthakal, S.V. Madathilparambil, P. McDonald, L.K. Olson, A. Kowluru // Biochem. Pharmacol. 2009 Jan 1; 77(1): 101-13. doi: 10.1016/j.bcp.2008.09.021.
31. Chen X.W. A Ral GAP complex links PI 3-kinase/Akt signa–ling to RalA activation in insulin action / X.W. Chen, D. Leto, T. Xiong et al. // Mol. Biol. Cell. 2011 Jan 1; 22(1): 141-52. doi: 10.1091/mbc.E10-08-0665.
32. Manyes L. Transcriptional profiling reveals functional links between RasGrf1 and Pttg1 in pancreatic beta cells / L. Manyes, M. Arribas, C. Gomez et al. // BMC Genomics. 2014 Nov 25; 15: 1019. doi: 10.1186/1471-2164-15-1019.
33. Yamamoto K. Overexpression of PACAP in transgenic mouse pancreatic beta-cells enhances insulin secretion and ameliorates streptozotocin-induced diabetes / K. Yamamoto, H. Hashimoto, S. Tomimoto et al. // Diabetes. 2003 May; 52(5): 1155-62. doi: 10.2337/diabetes.52.5.1155.
34. Harmar A.J. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activa–ting polypeptide: IUPHAR review 1 / A.J. Harmar, J. Fahrenkrug, I. Gozes et al. // Br. J. Pharmacol. 2012 May; 166(1): 4-17. doi: 10.1111/j.1476-5381.2012.01871.x.
35. Marzagalli R. Emerging Role of PACAP as a New Potential Therapeutic Target in Major Diabetes Complications / R. Marzagalli, S. Scuderi, F. Drago, J.A. Waschek, A. Castorina // Int. J. Endocrinol. 2015; 2015: 160928. doi: 10.1155/2015/160928.
36. Jamen F. PAC1 receptor-deficient mice display impaired insulinotropic response to glucose and reduced glucose tolerance / F. Jamen, K. Persson, G. Bertrand et al. // J. Clin. Invest. 2000 May; 105(9): 1307-15. doi: 10.1172/JCI9387.
37. Dickson J.L. A C-Peptide-Based Model of Pancreatic Insulin Secretion in Extremely Preterm Neonates in Intensive Care / J.L. Dickson, J. Alsweiler, C.A. Gunn // J. Diabetes Sci Technol. 2015 Aug 7. pii: 1932296815596175.
38. Ghorbani A., Shafiee-Nick R. Pathological consequences of C-peptide deficiency in insulin-dependent diabetes mellitus // World J. Diabetes. 2015 Feb 15; 6(1): 145-50. doi: 10.4239/wjd.v6.i1.145.
39. Boyraz M. Transient neonatal diabetes mellitus in a Tur–kish patient with three novel homozygous variants in the ZFP57 gene / M. Boyraz, K. Ulucan, N. Taşkın et al. // J. Clin. Res. Pediatr. Endocrinol. 2013; 5(2): 125-8. doi: 10.4274/Jcrpe.928.
40. Li X. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints/ X. Li, M. Ito, F. Zhou et al. // Dev Cell. 2008 Oct; 15(4): 547-57. doi: 10.1016/j.devcel.2008.08.014.
41. Quenneville S. In embryonic stem cells, ZFP57/KAP1 re–cognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions / S. Quenneville, G. Verde, A. Corsinotti et al. // Mol. Cell. 2011 Nov 4; 44(3): 361-72. doi: 10.1016/j.molcel.2011.08.032.
42. Zuo X. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain / X. Zuo, J. Sheng, H.T. Lau et al. // J. Biol. Chem. 2012 Jan 13; 287(3): 2107-18. doi: 10.1074/jbc.M111.322644.
43. Takikawa S. Human and mouse ZFP57 proteins are functionally interchangeable in maintaining genomic imprinting at multiple imprinted regions in mouse ES cells / S. Takikawa, X. Wang, C. Ray et al. // Epigenetics. 2013 Dec; 8(12): 1268-79. doi: 10.4161/epi.26544.
44. Baglivo I. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1 / Baglivo I., Esposito S., De Cesare L. et al. // FEBS Lett. 2013 May 21; 587(10): 1474-81. doi: 10.1016/j.febslet.2013.02.045.
45. Boonen S.E. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up / S.E. Boonen, D.J. Mackay, J.M. Hahnemann et al. // Diabetes Care. 2013 Mar; 36(3): 505-12. doi: 10.2337/dc12-0700.
1. Temple IK, Shield JP. 6q24 transient neonatal diabetes. Rev Endocr Metab Disord. 2010 Sep;11(3):199-204. doi: 10.1007/s11154-010-9150-4.
2. Rubio-Cabezas O, Ellard S. Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm Res Paediatr. 2013;80(3):137-46. doi: 10.1159/000354219
3. Kuraeva TL, Emelianov AO. [Clinical and genetic heterogeneity of neonatal diabetes]// Saharniy diabet. 2009;3:10-15. Russian.
4. Shield JP. Neonatal diabetes: how research unravelling the genetic puzzle has both widened our understanding of pancreatic development whilst improving children's quality of life. Horm Res. 2007;67(2):77-83. doi:10.1159/000096354
5. Hutchison JH, Keay AJ, Kerr MM. Congenital temporary diabetes mellitus. Br Med J. 1962 Aug 18;2(5302):436-40. PMID: 14450256
6. Stanik J et al. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab. 2007 Apr;92(4):1276-82. doi: http://dx.doi.org/10.1210/jc.2006-2490.
7. Wiedemann B et al. Incidence of neonatal diabetes in Austria-calculation based on the Austrian Diabetes Register. Pediatr Diabetes. 2010 Feb;11(1):18-23. doi: 10.1111/j.1399-5448.2009.00530.x.
8. Polak M, Shield J. Neonatal and very-early-onset diabetes mellitus. Semin Neonatol. 2004 Feb;9(1):59-65. doi: http://dx.doi.org/10.1016/S1084-2756(03)00064-2.
9. Flanagan SE et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007 Jul;56(7):1930-7. doi: 10.2337/db07-0043
10. Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008 May;29(3):265-91. doi: 10.1210/er.2007-0029.
11. Singerland AS et al. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia. 2009 Aug;52(8):1683-5. doi: 10.1007/s00125-009-1416-6.
12. Iafusco D et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012 Oct;49(5):405-8. doi: 10.1007/s00592-011-0331-8.
13. Milenkovic T et al. Transient neonatal diabetes mellitus in an infant with paternal uniparental disomy of chromosome 6 including heterodisomy for 6q24. J Pediatr Endocrinol Metab. 2006 Nov;19(11):1353-7. PMID: 17220064.
14. Prando C et al. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-gamma receptor 1 deficiency. Am J Med Genet A. 2010 Mar;152A(3):622-9. doi: 10.1002/ajmg.a.33291.
15. Suzuki S et al. Partial paternal uniparental disomy of chromosome 6 in monozygotic twins with transient neonatal diabetes mellitus and macroglossia. Clin Genet. 2010 Dec;78(6):580-4. doi: 10.1111/j.1399-0004.2010.01433.x.
16. Temple IK., Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002 Dec;39(12):872-5. doi: 10.1136/jmg.39.12.872.
17. Court F. et al. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum Mutat. 2013 Apr;34(4):595-602. doi: 10.1002/humu. 22276.
18. Strogantsev R et al. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015 May 30;16(1):112. doi: 10.1186/s13059-015-0672-7.
19. Temple IK, Mackay DJG, Docherty LE. Diabetes Mellitus, 6q24-Related Transient Neonatal. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2005 Oct 10 [updated 2015 Jan 15].PMID: 20301706.
20. Mitchell BD, Pollin TI. Genomic imprinting in diabetes. Genome Med. 2010 Aug 23;2(8):55. doi: 10.1186/gm176.
21. Mackay DJ et al A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet. 2006 Sep;120(2):262-9. doi: 10.1007/s00439-006-0205-2.
22. Rezwan FI et al. A statistical method for single sample analysis of HumanMethylation450 array data: genome-wide methylation analysis of patients with imprinting disorders. Clin Epigenetics. 2015 Apr 21;7(1):48. doi: 10.1186/s13148-015-0081-5.
23. Mackay DJ et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008 Aug;40(8):949-51. doi: 10.1038/ng.187.
24. Boonen SE et al. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur J Hum Genet. 2008 Apr;16(4):453-61. doi: 10.1038/sj.ejhg.5201993.
25. Rodríguez-Henche N et al. Transcription of the mouse PAC1 receptor gene: cell-specific expression and regulation by Zac1. Biochim Biophys Acta. 2002 Jun 7;1576(1-2):157-62. doi:10.1016/S0167-4781(02)00303-2.
26. Mackay DJ et al. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet. 2002 Feb;110(2):139-44. doi: 10.1007/s00439-001-0671-5.
27. Ma D et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004 Aug;114(3):339-48. doi: 10.1172/JCI200419876.
28. Hoffmann A, Spengler D. Transient neonatal diabetes mellitus gene Zac1 impairs insulin secretion in mice through Rasgrf1. Mol Cell Biol. 2012 Jul;32(13):2549-60. doi: 10.1128/MCB.06637-11.
29. Fernández-Medarde A, Santos E. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim Biophys Acta. 2011 Apr;1815(2):170-88. doi: 10.1016/j.bbcan.2010.11.001.
30. Veluthakal R et al. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem Pharmacol. 2009 Jan 1;77(1):101-13. doi: 10.1016/j.bcp.2008.09.021.
31. Chen XW et al. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell. 2011 Jan 1;22(1):141-52. doi: 10.1091/mbc.E10-08-0665.
32. Manyes L et al. Transcriptional profiling reveals functional links between RasGrf1 and Pttg1 in pancreatic beta cells. BMC Genomics. 2014 Nov 25;15:1019. doi: 10.1186/1471-2164-15-1019.
33. Yamamoto K et al. Overexpression of PACAP in transgenic mouse pancreatic beta-cells enhances insulin secretion and ameliorates streptozotocin-induced diabetes. Diabetes. 2003 May;52(5):1155-62. doi: 10.2337/diabetes.52.5.1155.
34. Harmar AJ et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012 May;166(1):4-17. doi: 10.1111/j.1476-5381.2012.01871.x.
35. Marzagalli R et al. Emerging Role of PACAP as a New Potential Therapeutic Target in Major Diabetes Complications. Int J Endocrinol. 2015;2015:160928. doi: 10.1155/2015/160928.
36. Jamen F et al. PAC1 receptor-deficient mice display impaired insulinotropic response to glucose and reduced glucose tolerance. J Clin Invest. 2000 May;105(9):1307-15. doi: 10.1172/JCI9387.
37. Dickson JL. A C-Peptide-Based Model of Pancreatic Insulin Secretion in Extremely Preterm Neonates in Intensive Care. J Diabetes Sci Technol. 2015 Aug 7. pii: 1932296815596175.
38. Ghorbani A, Shafiee-Nick R. Pathological consequences of C-peptide deficiency in insulin-dependent diabetes mellitus. World J Diabetes. 2015 Feb 15;6(1):145-50. doi: 10.4239/wjd.v6.i1.145.
39. Boyraz M et al. Transient neonatal diabetes mellitus in a Turkish patient with three novel homozygous variants in the ZFP57 gene. J Clin Res Pediatr Endocrinol. 2013;5(2):125-8. doi: 10.4274/Jcrpe.928.
40. Li X et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008 Oct;15(4):547-57. doi: 10.1016/j.devcel.2008.08.014.
41. Quenneville S et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011 Nov 4;44(3):361-72. doi: 10.1016/j.molcel.2011.08.032.
42. Zuo X et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2012 Jan 13;287(3):2107-18. doi: 10.1074/jbc.M111.322644.
43. Takikawa S et al. Human and mouse ZFP57 proteins are functionally interchangeable in maintaining genomic imprinting at multiple imprinted regions in mouse ES cells. Epigenetics. 2013 Dec;8(12):1268-79. doi: 10.4161/epi.26544.
44. Baglivo I et al. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1. FEBS Lett. 2013 May 21;587(10):1474-81. doi: 10.1016/j.febslet.2013.02.045.
45. Boonen SE et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up. Diabetes Care. 2013 Mar;36(3):505-12. doi: 10.2337/dc12-0700.