
Международный эндокринологический журнал 4 (60) 2014
Вернуться к номеру
Аутоиммунные полиэндокринные синдромы: классификация, клиника, диагностика, лечение
Авторы: Прилуцкий А.С., Прилуцкая О.А., Стрельченко Е.С. - Донецкий национальный медицинский университет им. М. Горького
Рубрики: Эндокринология, Иммунология
Разделы: Клинические исследования
Версия для печати
Аутоиммунные полиэндокринные синдромы являются актуальной проблемой современной медицины, поскольку включают в себя развитие нескольких эндокринопатий аутоиммунного характера у одного и того же пациента, сочетающихся с аутоиммунной неэндокринной патологией. В обзоре представлена сравнительная характеристика четырех типов аутоиммунных полиэндокринных синдромов, а также современные данные об их клинике, диагностике и лечении.
Автоімунні поліендокринні синдроми є актуальною проблемою сучасної медицини, оскільки містять у собі розвиток декількох ендокринопатій автоімунного характеру в одного й того ж пацієнта, що поєднуються з автоімунною неендокринною патологією. В огляді подано порівняльну характеристику чотирьох типів автоімунних поліендокринних синдромів, а також сучасні дані щодо їх клініки, діагностики та лікування.
Autoimmune polyendocrine syndromes are the actual problem of modern medicine, as include the development of several autoimmune endocrinopathies in the same patient, associated with autoimmune nonendocrine pathology. The review presents a comparative analysis of four types of autoimmune polyendocrine syndromes, as well as current data on their clinical picture, diagnosis and treatment.
аутоиммунные полиэндокринные синдромы, классификация, аутоиммунные заболевания, болезнь Аддисона.
автоімунні поліендокринні синдроми, класифікація, автоімунні захворювання, хвороба Аддисона.
autoimmune polyendocrine syndromes, classification, autoimmune diseases, Addison’s disease.
Статья опубликована на с. 13-20
Аутоиммунные полиэндокринные синдромы (АПС) являются очень актуальной проблемой современной медицины во всем мире, так как включают в себя комбинацию нескольких аутоиммунных заболеваний, что значительно ухудшает качество и уменьшает продолжительность жизни таких пациентов. Аутоиммунные полиэндокринные синдромы — группа заболеваний, при которых наблюдается одновременное или поэтапное развитие двух и более эндокринопатий аутоиммунного характера у одного и того же больного, которые совмещаются во многих случаях с аутоиммунной неэндокринной патологией [1]. В 1855 году Томас Аддисон обратил внимание на то, что у пациентов с идиопатической надпочечной недостаточностью могут иметь место другие аутоиммунные заболевания — пернициозная анемия и витилиго [2].
В 1908 году H. Claude и H. Gourgerot предположили общий патогенез нескольких аутоиммунных эндокринных заболеваний, описав его в статье под названием «Insufficance pluriglandulaire endocrinnienne» [3]. В 1926 году М. Schmidt показал связь между недостаточностью коры надпочечников и тиреоидитом [4]. Со временем в 1964 году H. Carpenter расширил синдром, который описал М. Schmidt, и включил в него инсулинзависимый сахарный диабет. Далее, после углубленного анализа больных с полиэндокринопатиями, Neufeld и R. Blizzard в 1980 году предложили назвать эту группу заболеваний аутоиммунными полиэндокринными синдромами. Также они разработали классификацию, которая основана на разделении АПС в зависимости от заболеваний, которые составляли каждый определенный тип АПС [6]. Были выделены четыре типа [6, 7] аутоиммунных полиэндокринных синдромов (табл. 1).
/14/14.jpg)
Аутоиммунный полиэндокринный синдром 1-го типа
АПС 1-го типа (АПС-1), называемый АPECED (autoimmune polyendocrinopathy, candidiasis, ectodermal-dystrophy) или MEDAC (multiple endocrine deficiency autoimmune candidiasis), характеризуется наличием хронического слизисто-кожного кандидоза, хронического гипопаратиреоза и аутоиммунной надпочечниковой недостаточности (болезнь Аддисона, АНН).
В основе АПС 1-го типа лежит дефект в аутоиммунно-регуляторном гене, который локализуется в 21-й хромосоме (21q22.3). Этот ген, получивший название AIRE (autoimmune regulator — аутоиммунный регулятор), кодирует белок AIRE, который, наиболее вероятно, является регулятором транскрипции [8, 9].
Этот синдром – довольно редкая патология с частотой распространения 1 : 100 000 населения и имеет моногенный аутосомно-рецессивный тип наследования. Относительно высокая частота наблюдается у финнов (1 : 25 000), среди иранских евреев (1 : 9000) и жителей Сардинии (1 : 14 000). Может немного чаще выявляться у мужчин (соотношение мужчин и женщин — 4 : 3) [15, 16].
В большинстве случаев АПС 1-го типа проявляется в детском или подростковом возрасте. Как правило, его первым симптомом является слизисто-кожный кандидоз, который развивается в первые 5 лет жизни. При этом наблюдается поражение слизистых оболочек полости рта, гениталий, а также кожи, ногтевых валиков и ногтей. До 10-летнего возраста обычно развивается гипопаратиреоз, который характеризуется нервно-мышечной гиперактивностью, гипотензией. Надпочечниковая недостаточность присоединяется в большей степени до 15-летнего возраста и проявляется в виде слабости, снижения веса, анорексии, тошноты, рвоты, абдоминальных болей, диареи, гипотонии, гипогликемии, кожной гиперпигментации. Также в ряде случаев могут выявляться различые психические и эмоциональные нарушения [17–19].
В состав АПС-1 могут также входить другие нарушения эндокринных органов, такие как: гипергонадотропный гипогонадизм, аутоиммунный тиреоидит, сахарный диабет 1-го типа (СД-1), реже — аутоиммунные поражения гипофиза [18, 20]. Нередко на первый план в клинической картине выходят аутоиммунные неэндокринные заболевания — витилиго, васкулиты. Со стороны желудочно-кишечного тракта также могут наблюдаться аутоиммунные атрофические гастриты, гепатиты с циррозом печени, интестинальная дисфункция и синдром мальабсорбции, пернициозная анемия [17, 18]. Очень часто в составе АПС 1-го типа присутствует алопеция и эктодермальная дистрофия, которая включает кератоконъюнктивиты, гипоплазию зубной эмали и точечные дефекты ногтей [15, 17, 21].
Диагностика синдрома основана на выявлении классической триады, характерной для АПС-1: слизисто-кожный кандидоз, гипопаратиреоз, первичная хроническая надпочечниковая недостаточность. Для установления диагноза достаточно наличия у больного двух из трех вышеперечисленных заболеваний. Слизисто-кожный кандидоз характеризуется наличием селективного дефицита Т-клеточного уровня на фоне нормального В-клеточного ответа. Как правило, наблюдается высокий титр антител к Candida аlbicans [10]. При хроническом гипопаратиреозе выявляются гипокалиемия, гиперфосфатемия, снижение уровня кальция и определяются аутоантитела к ткани паращитовидной железы и Са2+-рецепторам [25]. Для диагностики надпочечниковой недостаточности проводят МРТ- или КТ-исследование надпочечников, результаты которых могут свидетельствовать о гипо- или атрофии органа. Часто выявляются аутоантитела к надпочечникам. В анализе крови может выявляться эозинофилия с лимфоцитозом, микро- или макроцитарная анемия. Для надпочечниковой недостаточности характерны гипонатриемия, гипохлоремия, гиперкалиемия, снижение осмолярности плазмы [26, 27].
Лечение слизисто-кожного кандидоза проводят при помощи итраконазола, амфотерицина и др. Для заместительной терапии гипопаратиреоза используют препараты кальция, витамина D и др. При надпочечниковой недостаточности используют глюко- и минералокортикоиды. Следует иметь в виду, что при назначении глюкокортикоидов их передозировка может вызывать декомпенсацию гипопаратиреоза и спровоцировать развитие гипокальциемии [17, 18, 21].
Аутоиммунный полиэндокринный синдром 2–го типа
АПС 2-го типа (АПС-2) характеризуется поражением эндокринных желез с развитием надпочечниковой недостаточности (первичного гипокортицизма), аутоиммунного тиреоидита и СД 1-го типа. Эти проявления нередко сопровождают первичный гипогонадизм, миастению гравис, витилиго, алопецию, аутоиммунный гепатит, хронический атрофический гастрит, пернициозную анемию, гипофизит. В состав АПС 2-го типа также могут входить аутоиммунная тромбоцитопеническая пурпура, идиопатический несахарный диабет с аутоантителами к вазопрессинпродуцирующим клеткам, изолированный дефицит адренокортикоидного гормона, опухоли гипофиза, склеродермия и др.
АПС-2 сам по себе является достаточно редким (1,4–2,0 случая на 100 000 населения) и манифестирует, как правило, у женщин средних лет (средний возраст появления заболевания — 35 лет) [28, 29]. Отмечено, что АПС 2-го типа имеет полигенное наследование, в ряде случаев доминантное с неполной пенетрантностью и ассоциирован, по современным данным, главным образом с HLA [31, 32]. Начало заболевания чаще проявляется клиникой первичной хронической недостаточности коры надпочечников. Аутоиммунный тиреоидит и СД 1-го типа присоединяются в среднем через 7 лет, хотя разрыв между началом заболеваний может быть до 20 лет [27, 35].
Различают 2А- и 2В-подтип АПС 2-го типа. Наиболее частым вариантом АПС 2-го типа является синдром Шмидта (2А-подтип), при котором наблюдается надпочечниковая недостаточность, которая ассоциируется с аутоиммунным тиреоидитом. Синдром Карпентера, или 2В-подтип, характеризуется комбинацией надпочечниковой недостаточности и СД 1-го типа. При наличии синдрома Шмидта — Карпентера надпочечниковая недостаточность сочетается одновременно с аутоиммунным тиреоидитом и СД 1-го типа.
Диагностика основана на выявлении основных эндокринопатий, характерных для АПС-2, выявлении аутоантител к определенным аутоантигенам в сыворотке крови [1, 28, 36]. Дифференциальную диагностику следует проводить с изолированными аутоиммунными эндокринопатиями. При тяжелом тиреотоксикозе, при болезни Грейвса у пациента могут быть явления относительной надпочечниковой недостаточности (легкая гиперпигментация, гипотония и т.д.), которую необходимо дифференцировать от истинной [37, 38].
Лечение пациентов проводится с использованием комбинированной заместительной терапии при недостаточности нескольких эндокринных желез. Лечение каждого компонента аутоиммунного полиэндокринного синдрома проводится так, как если бы каждый синдром, который входит в состав АПС-2, был отдельным заболеванием [28, 29].
Аутоиммунный полиэндокринный синдром 3-го типа
Аутоиммунный полиэндокринный синдром 3-го типа (АПС-3) определяется авторами [6, 41, 42] как комбинация аутоиммунных тиреопатий (болезнь Хашимото, идиопатическая микседема, бессимптомный аутоиммунный тиреоидит, болезнь Грейвса и др.) с другими заболеваниями эндокринных органов, гастроинтестинальными аутоиммунными нарушениями, нарушениями со стороны кожи, системы гемопоэза и нервной системы, васкулитами и др.
Согласно современным данным, АПС 3-го типа, как и АПС 2-го типа, ассоциирован с HLA-генами II класса. Так, НLA-DRB1*04/-DQA1*0301/-DQB1*0302 связаны с СД-1, HLA-DQB1*0301 часто ассоциирован с аутоиммунным тиреоидитом, HLA-DRB1*0401 и -DQB1*0301 — с алопецией. Вместе с тем установлено, что существуют также не-HLA-гены, связанные с возникновением АПС-3, которые еще необходимо идентифицировать. АПС 3-го типа, вероятно, имеет аутосомно-доминантное наследование с неполной пенетрантностью. При семейных формах может проявляться в нескольких поколениях. Встречается чаще у женщин в возрасте 30–40 лет [43–45]. Распространенность АПС 3-го типа составляет приблизительно 5–7,5 случая на 100 000 населения [46, 47].
Аутоиммунный тиреоидит является основной составляющей у АПС 3-го типа. К другим заболеваниям эндокринных органов при АПС 3-го типа относят в первую очередь СД-1, болезнь Хирата (аутоиммунный инсулиновый синдром), лимфоцитарный гипофизит, первичный гипогонадизм и др. В последнее время доказано, что недостаточность соматотропного гормона также может быть одним из компонентов АПС-3 [48]. Аутоиммунные гастроинтестинальные поражения могут проявляться при этом синдроме в виде хронического атрофического гастрита, целиакии, аутоиммунного гепатита, первичного билиарного цирроза, склерозирующего холангита и др. К поражениям кожи относятся витилиго и алопеция, к поражению системы гемопоэза — аутоиммунная тромбоцитопения, аутоиммунная гемолитическая анемия, антифосфолипидный синдром. При нарушениях со стороны нервной системы при АПС-3 могут наблюдаться такие заболевания, как миастения гравис, синдром Штиффмана (синдром ригидного человека) и рассеянный склероз. В состав АПС-3 могут также входить диффузные заболевания соединительной ткани: системная красная волчанка, ревматоидный артрит, синдром Шегрена, склеродермия и др. [49–53].
В 1980 г. M. Neufeld и R. Blizzard [6] предложили первую классификацию АПС, где АПС 3-го типа под-разделяется на три категории: АПС-3А — аутоиммунный тиреоидит в сочетании с аутоиммунным сахарным диабетом с другими аутоиммунными заболеваниями или без них; АПС-3В — аутоиммунный тиреоидит с пернициозной анемией, АПС-3С — аутоиммунный тиреоидит, витилиго и/или алопеция.
Более новая классификация, которую предложил C. Betterle et al. в 2001 году, делит АПС 3-го типа на 4 подгруппы (табл. 2) [54].
/15/15.jpg)
Согласно этой классификации, к АПС-3 относятся 3А-подтип (аутоиммунный тиреоидит и упомянутые в табл. 2 другие аутоиммунные эндокринные заболевания); 3В-подтип (аутоиммунный тиреоидит и гастроинтестинальные поражения); 3С-подтип (аутоиммунный тиреоидит и поражения кожи, системы гемостаза и нервной системы); 3D-подтип (аутоиммунный тиреоидит и диффузные заболевания соединительной ткани). Однако, согласно определению, аутоиммунный полиэндокринный синдром — это заболевание, которое включает два и более поражения со стороны эндокринных органов в комбинации с другими аутоиммунными заболеваниями [1]. Поэтому разделение АПС 3-го типа таким образом, по нашему мнению, не совсем уместно. Более того, в настоящее время в литературе описан так называемый множественный аутоиммунный синдром (multiple autoimmune syndrome). Авторами указывается, что наличие у одного и того же пациента нескольких аутоиммунных заболеваний позволяет диагностировать у него множественный аутоиммунный синдром [55–57].
В свою очередь, он подразделен на три группы, в каждой из которых может объединяться несколько аутоиммунных неэндокринных, а также, возможно, одно эндокринное аутоиммунное заболевание [58]. Поэтому, исходя из всего вышеперечисленного, комбинация одного аутоиммунного эндокринного заболевания с другими неэндокринными аутоиммунными заболеваниями корректнее отнести в одну из групп множественного аутоиммунного синдрома. Учитывая современные данные о возможных ассоциациях аутоиммунных заболеваний щитовидной железы, по нашему мнению, АПС 3-го типа должен включать 3А-подтип — аутоиммунный тиреоидит и аутоиммунные эндокринные заболевания поджелудочной железы, а подтипы 3В, 3С и 3D — аутоиммунный тиреоидит в сочетании с другими аутоиммунными эндокринопатиями (гипофизит, гипогонадизм и др.).
Диагностика АПС 3-го типа также основана на выявлении характерных клинических проявлений, специфических аутоантител в сыворотке крови, наличия лимфоцитарной инфильтрации пораженных органов [41].
Лечение зависит от того, какие органы включены в патологический процесс. Показан мониторинг структуры и функции эндокринных органов, длительная гормональная заместительная терапия. Таким пациентам необходимы пожизненное наблюдение и обследование на предмет выявления других компонентов синдрома. Следует заметить, что длительный мониторинг возникновения новых и прогрессирования существующих аутоиммунных заболеваний показан всем больным с наличием аутоиммунной патологии [6]. При этом необходимо учитывать, что у 25 % пациентов в течение жизни наблюдается присоединение дополнительных аутоиммунных заболеваний.
Аутоиммунный полиэндокринный синдром 4-го типа
АПС 4-го типа (АПС-4) — редкий синдром, который характеризуется сочетанием аутоиммунных заболеваний, не вошедших в предыдущие категории АПС [6, 7]. В состав АПС 4-го типа могут входить, например, болезнь Аддисона в сочетании с гипогонадизмом, гипофизитом, атрофическим гастритом, пернициозной анемией, целиакией и др. При этом отсутствуют другие компоненты АПС-1, то есть нет ни хронического кандидоза, ни гипопаратиреоза (при наличии аутоиммунной недостаточности надпочечников), а также отсутствуют компоненты АПС-2, -3 (аутоиммунные тиреоидиты; СД-1 при основном заболевании — болезни Аддисона) [7, в нашей модификации].
Характеристика АПС 4-го типа, по нашему мнению, может быть представлена следующим образом (табл. 3).
/16/16.jpg)
Диагностика АПС 4-го типа также основана на выявлении характерных клинических проявлений, определении аутоантител в сыворотке крови. Необходимо указать, что АПС 4-го типа является наименее изученным среди всех АПС. До настоящего времени остаются неизученными этиология, частота возникновения, возраст манифестации, тип наследования, генетическая предрасположенность. Следует отметить, что АПС 4-го типа может включать, по нашему мнению, комбинацию (как основное заболевание может быть одно из других эндокринных заболеваний, кроме аутоиммунных заболеваний щитовидной железы) прочих аутоиммунных заболеваний эндокринной системы. Сочетание же двух или более аутоиммунных заболеваний неэндокринной системы целесообразно отнести к группам полииммунных заболеваний, множественных аутоиммунных синдромов.
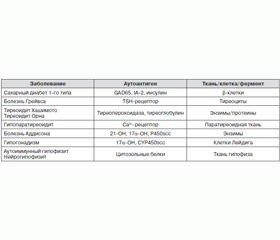
Анализируя все вышеизложенное, можно отметить, что в выделенных четырех типах АПС просматривается ряд общих признаков, характерных для аутоиммунных заболеваний. В первую очередь необходимо указать наличие у всех больных аутоантител к антигенам, которые коррелируют с клиническими проявлениями заболеваний разных эндокринных органов, входящих в состав АПС (табл. 4). Следует отметить, что данные антитела могут появляться намного ранее развития клинических симптомов соответствующих заболеваний. Периодическое тестирование их целесообразно у всех пациентов с наличием хотя бы одного аутоиммунного заболевания с целью определения риска возникновения клинических проявлений других, новых аутоиммунных болезней.
/17/17.jpg)
Ниже представлена сравнительная характеристика аутоиммунных полиэндокринных синдромов (табл. 5). Среди взрослых со 2-м и 3-м типом АПС заболевания наблюдаются преимущественно у женщин средних лет.
АПС 1-го типа характеризуется манифестацией симптомов в детском возрасте, болеют чаще мужчины. В то же время следует отметить наличие у большинства типов АПС доказанного или предполагаемого полигенного типа наследования, в ряде случаев с неполной пенетрантностью. Однако АПС 1-го типа имеет моногенный, аутосомно-рецессивный тип наследования, при котором наблюдается дефект гена AIRE в 21-й хромосоме. Отдельными исследованиями у ряда пациентов с АПС 2-го типа были выявлены нарушения в 6-й хромосоме. Точной локализации генных поражений для АПС 3-го и 4-го типов в настоящее время еще не найдено. Показано, что у большинства из синдромов есть ассоциации с аллоантигенами HLA. Обязательными эндокринными синдромами для АПС 1-го типа являются аутоиммунная надпочечниковая недостаточность, гипопаратиреоз, для АПС 2-го типа — аутоиммунная надпочечниковая недостаточность, аутоиммунный тиреоидит и/или СД 1-го типа, для АПС 3-го типа — аутоиммунный тиреоидит и другие заболевания эндокринных органов (кроме недостаточности надпочечников), для АПС 4-го типа — надпочечниковая недостаточность, СД 1-го типа и другие заболевания эндокринных органов. Следует отметить, что среди неэндокринных составляющих обязательным признаком АПС 1-го типа является кандидоз-эктодермальная дистрофия. Другими же аутоиммунными заболеваниями, которые входят в состав АПС практически всех типов, являются, как правило, витилиго, алопеция, васкулиты, гастрит, гепатит, интестинальная дисфункция, пернициозная анемия, синдром мальабсорбции, антифосфолипидный синдром, миастения гравис, системная красная волчанка, ревматоидный артрит, синдром Шегрена, склеродермия и др.
1. Dittmar M. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up / M. Dittmar, G.J. Kahaly // Journal of Clinical Endocrinology and Metabolism. — 2003. — Vol. 88, № 7. — Р. 2983–2992.
2. Addison T. 1855 on the constitutional and local effects of disease of the suprarenal capsules. In a collection of the published writing of the late / Thomas Addison. — London: New Sydenham Society, 1868; Reprinted in Medical Classics, 1937. — Р. 244–293.
3. Claude H. Insuffisance pluriglandulaire endocrinienne / H. Claude, H. Gourgerot // Journal Physiol. Pathol. Gen. — 1908. — Vol. 10. — P. 469–80.
4. Schmidt M.B. Eine biglandulare Erkrankung (Nebennieren und Schilddrüse) bei Morbus Addisonii / M.B. Schmidt // Verh. Dtsch. Ges. Pathol. — 1926. — Vol. 21. — Р. 212–221.
5. Carpenter C.J. Schmidt’s syndrome (thyroid and adrenal insufficiency): a review of the literature and a report of fifteen new cases including ten instances of coexistent diabetes Mellitus / C.J. Carpenter, N. Solomon, Silverberg et al. // Medicine. — 1964. — Vol. 43. — P. 153–180.
6. Neufeld M. Polyglandular autoimmune diseases / M. Neufeld, R.M. Blizzard // Pediatr Ann. — 1980. — Vol. 9, № 4. — P. 154–162.
7. Betterle C. Update on autoimmune polyendocrine syndromes (APS) / C. Betterle, R. Zanchetta // Acta Biomedica. — 2003. — Vol. 74. — P. 9–33.
8. Finnish–German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD type zinc–finger domains // Nature Genetics. — 1997. — Vol. 17. — P. 399–403.
9. Anderson M.S. Projection of an immunological self shadow within the thymus by the aire protein / M.S. Anderson, E.S.Venanzi, L. Klein [et al.] // Science. — 2002. — Vol. 298. — P. 1395–1401.
10. Conti H.R. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis / H.R. Conti, F. Shen, N. Nayyar [et al.] // Journal of Experimental Medicine. — 2009. — Vol. 206, № 2. — P. 299–311.
11. Puel A. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I / A. Puel, R. Doffinger, A. Natividad [et al.] // Journal of Experimental Medicine. — 2010. — Vol. 207, № 2. — P. 291–297.
12. Kumar P.G. Population genetics and functions of the autoimmune regulator (AIRE) / P.G. Kumar, M. Laloraya, J.X. She // Endocrinology and Metabolism Clinics of North America. — 2002. — Vol. 31. — P. 321–338.
13. Pearce S.H. A common and recurrent 13 — bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1 / S.H. Pearce, T. Cheetham, H. Imrie [et al.] // American Journal of Human Genetics. — 1998. — Vol. 63. — P. 1675–1684.
14. Scott H.S. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins / H.S. Scott, M. Heino, P. Peterson [et al.] // Molecular Endocrinology. — 1998. — Vol. 12. — P. 1112–1119.
15. Rosatelli M.C. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients / M.C. Rosatelli, A. Meloni, M. Devoto [et al.] // Human genetics. — 1998. — Vol. 103, № 4. — P. 428–434.
16. Zlotogora J. Polyglandular autoimmune syndrome type I among Iranian Jews / J. Zlotogora, M.S. Shapiro // Journal of Medical Genetics. — 1992. — Vol. 29, № 11. — P. 824–826.
17. Betterle C. Clinical review 93: autoimmune polyglandular syndrome type 1 / C. Betterle, N.A. Greggio, M. Volpato // Journal of Clinical Endocrinology & Metabolism. — 1998. — Vol. 83, № 4. — P. 1049–1055.
18. Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy / J. Perheentupa // Journal of Clinical Endocrinology & Metabolism. — 2006. — Vol. 91, № 8. — P. 2843–2850.
19. Meager A. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1 / A. Meager, K. Visvalingam, P. Peterson [et al.] // PLOS Medicine. — 2006. — Vol. 3, № 7. — P. 289.
20. Soderbergh A. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I / A. Soderbergh, A.G. Myhre, O. Ekwall [et al.] // Journal of Clinical Endocrinology & Metabolism. — 2004. — Vol. 89(2). — P. 557–562.
21. Ahonen P. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients / P. Ahonen, S. Myllarniemi, I. Sipila, J. Perheentupa // New England Journal of Medicine. — 1990. — Vol. 322, № 26. — P. 1829–1836.
22. Ulinski T. Autoimmune polyendocrinopathy — candidiasis — ectodermal dystrophy syndrome with renal failure: impact of posttransplant immunosuppression on disease activity / T. Ulinski, L. Perrin, M. Morris [et al.] // Journal of Clinical Endocrinology & Metabolism. — 2006. — Vol. 91, № 1. — P. 192–195.
23. Garcha-Hernández F.J. Autoimmune polyglandular syndrome and pulmonary arterial hypertension / F.J. Garcha-Hernández, C. Ocaсa-Medina, R. González-Leуn [et al.] // European Respiratory Journal. — 2006. — Vol. 27, № 3. — P. 657–658.
24. Alimohammadi M. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen / M. Alimohammadi, N. Dubois, F. Skoldberg, A. Hallgren, I. Tardivel, H. Hedstrand [et al.] // Proceedings of the National Academy of Sciences of the United States of America. — 2009. — Vol. 106, № 11. — P. 4396–4401.
25. Myhre A.G. Autoimmune polyendocrine syndrome type 1 (APS I) in Norway / A.G. Myhre, M. Halonen, P. Eskelin, O. Ekwall [et al.] // Clin. Endocrinol. — 2001. — Vol. 54. — P. 211–217.
26. Oelkers W. Adrenal insufficiency / W. Oelkers // The New England Journal of Medicine. — 1996. — Vol. 335. — P. 1206–1212.
27. Ten S. Clinical Review 130: Addison’s disease / S. Ten, M. New, N. MacLaren // Journal of Clinical Endocrinology & Metabolism. — 2001. — Vol. 86. — P. 2909–2922.
28. Betterle C. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction / C. Betterle, C. Dal Pra, F. Mantero, R. Zanchetta // Endocrine Reviews. — 2002. — Vol. 23. — P. 327–364.
29. Schatz D.A. Autoimmune polyglandular syndrome. II: Clinical syndrome and treatment / D.A. Schatz, W.E. Winter // Endocrinol. Metab. Clin. North. Am. — 2002. — Vol. 31. — P. 339–352.
30. Michels A.W. Autoimmune polyendocrine syndrome type 1 (APS–1) as a model for understanding autoimmune polyendocrine syndrome type 2 (APS-2) / A.W. Michels, G.S. Eisenbarth // Journal of Internal Medicine. — 2009. — Vol. 265. — P. 530–540.
31. Wallaschofski H. HLADQA1*0301–associated susceptibility for autoimmune polyglandular syndrome type II and III / H. Wallaschofski, A. Meyer, U. Tuschy, T. Lohmann // Hormone and Metabolic Research. — 2003. — Vol. 35. — P. 120–124.
32. Skordis N. Immunogenetics of autoimmune polyglandular syndromes / Skordis N., Maclaren N. — Chapter 15th // Immunogenetics of Endocrine Disorders. — N.R. Farid (ed.), R. Liss Alan, Inc. — New York, 1988. — P. 373–399.
33. Graves L. Addisonian crisis precipitated by thyroxine therapy: a complication of type 2 autoimmune polyglandular syndrome / L. Graves, R.M. Klein, A.D. Walling // Southern Medical Journal. — 2003. — Vol. 96. — P. 824–827.
34. Robles DT. The genetics of autoimmune polyendocrine syndrome type II / D.T. Robles, P.R. Fain, P.A. Gottleib, G.S. Eisenbarth // Endocrinology and Metabolism Clinics of North America. — 2002. — Vol. 31. — P. 353–368.
35. Muir A. Autoimmune diseases of the adrenal glands, parathyroid glands, gonads, and hypothalamic — pituitary axis / A. Muir, N.K. Maclaren // Endocrinology and Metabolism Clinics of North America. — 1991. — Vol. 20. — P. 619–644.
36. Krohn K. Identification by molecular cloning of an autoantigen associated with Addison’ disease as steroid 17-a-hydroxylase / K. Krohn // Lancet. — 1992. — P. 339–770.
37. Weetman A.P. Graves’ Disaese / A.P. Weetman // The New England Journal of Medicine. — 2000. — Vol. 34. — P. 1236–1248.
38. Dayan C.M. Chronic autoimmune thyroiditis / C.M. Dayan, G.H. Daniels // The New England Journal of Medicine. — 1996. — Vol. 335. — P. 99.
39. Eisenbarth G.S. Autoimmune polyendocrine syndromes / G.S. Eisenbarth, P.A. Gottlieb // The New England Journal of Medicine. — 2004. — Vol. 350. — P. 2068–2079.
40. Gumieniak O. Schmidt’s syndrome and severe hyponatremia: report of an unusual case and review of the related literature / O. Gumieniak, A.P. Farwell // Endocrine Practice. — 2003. — Vol. 9. — P. 384–388.
41. Muir A. Advances in the genetic and immunology of autoimmune polyglandular syndrome II/III and their clinical applications / A. Muir, J.X. She // Annals of Internal Medicine. — 1999. — Vol. 150. — P. 301–12.
42. Kahaly G.J. Polyglandular autoimmune syndromes / G.J. Kahaly // European Journal of Endocrinology. — 2009. — Vol. 161, № 1. — P. 11–20.
43. Villano M.J., Huber A.K., Greenberg D.A. et al. Autoimmune thyroiditis and diabetes: dissecting the joint genetic susceptibility in a large cohort of multiplex families / M.J. Villano, A.K. Huber, D.A. Greenberg [et al.] // Journal of Clinical Endocrinology & Metabolism. — 2009. — Vol. 94, № 4. — P. 1458–1466.
44. Huber A. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms / A. Huber, F. Menconi, S. Corathers [et al.] // Endocrine Reviews. — 2008. — Vol. 29, № 6. — P. 697–725.
45. Fourati H. HLA–DRB1/DQB1 susceptibility for autoimmune polyglandular syndrome type II and III in south of Tunisia / H. Fourati, N. Mahfoudh, O. Abida [et al.] // Annales d’Endocrinologie. — 2011. — 72, № 3. — P. 232–238.
46. Anderson M.S. Update in endocrine autoimmunity / M.S. Anderson // Journal of Clinical Endocrinology & Metabolism. — 2008. — Vol. 93. — P. 3663–3670.
47. Driessche A. Van den. Type 1 diabetes and autoimmune polyglandular syndrome: a clinical review / A. Van den Driessche, V. Eenkhoorn, L. Van Gaal // The Netherlands Journal of Medicine. — 2009. — Vol. 67, № 11. — P. 376–87.
48. Quintos J.B. Autoimmune polyglandular syndrome Type 3 and growth hormone deficiency / J.B. Quintos, M. Grover, C.M. Boney, M. Salas [et al.] // Pediatric Diabetes. — 2010. — Vol. 11, № 6. — P. 438–442.
49. Ugur-Altun B. Autoimmune polyglandular syndrome type III in monozygotic twins: a case report / B. Ugur-Altun, E. Arikan, S. Guldiken, M. Kara // Acta Clinica Belgica. — 2004. — Vol. 59, № 4. — P. 225–228.
50. Shimomura H. A rare case of autoimmune polyglandular syndrome type 3 / H. Shimomura, Y. Nakase, H. Furuta, M. Nishi [et al.] // Diabetes Research and Clinical Practice. — 2003. — Vol. 61, № 2. — P. 103–108.
51. Oki K. A case of polyglandular autoimmune syndrome type III complicated with autoimmune hepatitis / K. Oki, K. Yamane, J. Koide, K. Mandai [et al.] // Endocrine Journal. — 2006. — Vol. 53, № 5. — P. 705–709.
52. Noriko O. Autoimmune Polyglandular Syndrome Type III Associated with Slowly Progressive Type 1 Diabetes Mellitus, Chronic Thyroiditis, Pernicious Anemia and Idiopathic Thrombocytopenic Purpura: A Case Report / O. Noriko, T. Junko, Y. Yuko [et al.] // Journal of the Japan Diabetic Society. — 2006. — Vol. 49. — P. 723–729.
53. Lubinska M. Acquired von Willebrand’s syndrome in a patient with severe primary hypothyroidism associated with myasthenia gravis in the course of autoimmune polyglandular syndrome type 3 / M. Lubinska, R. Swiatkowska-Stodulska, E. Kazimierska // Haemophilia. — 2007. — Vol. 13, № 5. — P. 675–676.
54. Betterle C. Generalità sulle Malattie Autoimmuni / C. Betterle, F. Presotto, R. Zanchetta // Le Malattie Autoimmuni. — Piccin (Ed.) Padova. — 2001. — P. 9–36.
55. Mohan M.P. Multiple autoimmune syndrome / M.P. Mohan, T.C. Ramesh // Indian Journal of Dermatology, Venereology and Leprology. — 2003. — Vol. 69. — P. 298–299.
56. Ai J. Autoimmune thyroid diseases: Etiology, pathogenesis, and dermatologic manifestations / Ai J., J.M. Leonhardt, W.R. Heymann // Journal of the American Academy of Dermatology. — 2003. — Vol. 48. — P. 641–659.
57. Humbert P. Les syndromes autoimmuns multiples / P. Humbert, J.L. Dupont // Annals of Internal Medicine. — 1988. — Vol. 139. — P. 159–68.
58. Cojocaru M. Multiple autoimmune syndrome / M. Cojocaru, I. Silosi // Journal of clinical medicine. — Vol. 5, № 2. — 2010. — P. 132–134.