
Журнал «Здоровье ребенка» 2 (23) 2010
Вернуться к номеру
Клиническое наблюдение ребенка, больного лепречаунизмом
Авторы: Феклин В.А., Сиренко Т.В., Здыбская О.П., Романова А.С., Синдеева Н.Т., Гайдамака Л.Н., Харьковский национальный медицинский университет, КУОЗ Областная детская клиническая больница № 1
Рубрики: Педиатрия/Неонатология
Версия для печати
В статье описано редкое клиническое наблюдение — лепречаунизм у ребенка раннего возраста.
Дети, гипотрофия, лепречаунизм.
Цель сообщения: ознакомить врачей с особенностями диагностики редкой патологии у детей — лепречаунизма.
Ребенок Д., мальчик, 5 мес., поступил 26.11.2008 г. в отделение детей младшего возраста КУОЗ Областная детская клиническая больница № 1 г . Харькова.
Жалобы матери при госпитализации на снижение аппетита, вялое сосание, затруднение глотания, отсутствие прибавки массы тела, задержку психомоторного развития. Родители считают ребенка больным с момента рождения, когда появились трудности с кормлением, отсутствие прибавки массы тела.
Из анамнеза жизни известно, что ребенок родился от IV беременности ( I беременность — мальчик, 12 лет; II и III беременности — медицинские аборты). Настоящая беременность протекала на фоне хронического пиелонефрита, материнско-плодовой инфекции. Родители и старший брат ребенка здоровы, семейный анамнез по сахарному диабету не отягощен.
При УЗИ в сроке беременности 20 недель выявлены черепно-лицевые дисморфии, укорочение длинных трубчатых костей, признаки материнско-плодовой инфекции (повышение эхогенности плаценты, многоводие).
Роды путем кесарева сечения в сроке беременности 36 недель. Масса ребенка при рождении 3100 г , длина тела 49 см , окружность головы 35 см , окружность груди 33 см . Оценка по шкале Апгар 6–8 баллов, состояние ребенка после рождения средней степени тяжести. На третьи сутки жизни у ребенка была диагностирована внутриутробная пневмония, ребенок переведен в отделение патологии новорожденных Харьковского городского родильного дома с перинатальным центром, где находился на лечении в течение 22 дней. Диагноз при выписке: внутриутробная двусторонняя очагово-сливная пневмония, синдром дыхательной недостаточности, кандидоз слизистой оболочки полости рта, перинатальное поражение ЦНС, синдром ликворо-динамических нарушений, синдром персистирующего фетального кровообращения — функционирующее овальное окно. Находился на смешанном вскармливании, докармливался смесью «Малютка», не привит.
После выписки из стационара у ребенка не увеличивалась масса тела, которая оставалась такой же, как при рождении, в течение 5 месяцев. Отмечалась задержка психомоторного развития. Ребенок направлен на стационарное лечение.
При поступлении состояние ребенка расценено как тяжелое. Тяжесть состояния была обусловлена гипотрофией, метаболическими расстройствами, неврологическими нарушениями. Дефицит массы тела составил 40 % (фактическая масса — 3100 г , долженствующая — 5200 г ). Дефицит длины тела — 17 % (фактическая длина — 50 см , долженствующая — 60,5 см ). Ребенок был вялым, адинамичным. Сосал из рожка вяло, высасывал по 30–40 мл каждый час, долго держал смесь во рту, отмечалось затруднение глотания, периодически срыгивал. После кормления быстро засыпал, периодически был беспокойным. Взгляд на предметах не фиксировал, голову не держал. Отмечалась резкая мышечная гипотония. Объем спонтанной двигательной активности был снижен.
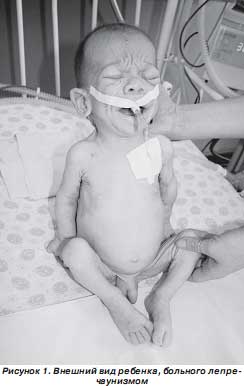
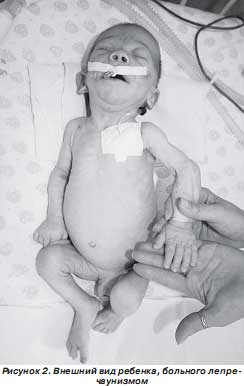
При осмотре обращали на себя внимание особенности внешнего вида ребенка (рис. 1, 2): черты лица гротескные, выпуклые глаза, плоская переносица, большой рот с толстыми губами, расширенный кончик носа, уши большие, низко расположенные, готическое небо, половой член увеличен. Кожные покровы сухие, бледные, эластичность кожи снижена. Подкожно-жировой слой отсутствует на животе, туловище, на конечностях, резко истончен на лице, отмечается образование дополнительных складок кожи на конечностях и туловище, лоб покрыт морщинами. Лицо ребенка напоминает лицо старика. Носогубная складка глубокая, челюсти и скулы выдаются, подбородок заострен, щеки западают, ушные раковины увеличены, распложены низко. Отмечается диспропорциональное телосложение ребенка — удлинение туловища и укорочение конечностей. Необычно большие кисти и стопы. Выражена мышечная гипотония. Имеется правосторонняя паховая грыжа, неущемленная. Задержка психомоторного развития: не фиксирует взгляд, не дает реакции оживления на контакт с матерью, не улыбается, не сосет, не глотает, не держит голову, не переворачивается со спины на живот и с живота на спину.
Грудная клетка — нижняя апертура расширена. Над легкими перкуторно легочный тон, аускультативно дыхание жесткое над симметричными участками. Границы сердца перкуторно не расширены (правая — по правой парастернальной линии, верхняя — на уровне второго ребра, левая — на 2 см снаружи от левой среднеключичной линии). Аускультативно тоны сердца ритмичные, несколько приглушены, систолический шум во второй точке аускультации, проводится на спину между лопатками.
Живот доступен пальпации, растянут, вздут, отмечается расхождение прямых мышц живота. Печень пальпируется на 3 см ниже края реберной дуги, селезенка не пальпируется.
Физиологические отправления: склонность к запорам, стул после клизмы, мочеиспускание свободное, отмечалось снижение суточного диуреза.
Результаты лабораторно-инструментального обследования ребенка
Клинический анализ крови: эр. — 3,7 х 10 12 /л, Hb — 94 г/л, ЦП — 0,74, тр. — 170 х 10 9 /л, Ht — 28 %, лейк. — 11,8 ´ 10 9 /л, э. — 4 %, п. — 4 %, с. — 65 %, л. — 20 %, м. — 7 %, СОЭ — 3 мм/час. Анизохромия, анизоцитоз. Токсическая зернистость лейкоцитов.
Клинический анализ мочи: к-во — 6,0 мл, цвет — светло-желтый, прозрачная, удельный вес — 1009, реакция — слабощелочная; белок, сахар, ацетон — не обнаружены. Микроскопия осадка: эпителий переходный — местами, лейкоциты — 2–4 в п/з; эритроциты: неизмененные — единичные, измененные — 0–1 в п/з; слизь — умеренное к-во, соли фосфаты — значительное количество.
Копрограмма: патологических изменений не выявлено.
Биохимический анализ крови: общий белок — 43,2 г/л, креатинин — 0,045 ммоль/л, мочевина — 6,94 ммоль/л, билирубин общий — 8,0 мкмоль/л, прямой — 1,5 мкмоль/л, непрямой — 6,5 мкмоль/л, АлАТ — 0,4 ммоль(ч·л), АсАТ — 0,25 ммоль(ч·л), щелочная фосфатаза — 143,3 ед/л, a -амилаза — 6,8 мг/(с·л), b -липопротеиды — 62,6 усл.ед., холестерин — 3,1 ммоль/л.
Обращала на себя внимание стойкая гипогликемия: 2,2; 1,7; 1,8; 2,4; 2,1; 2,0 ммоль/л.
Гормоны коры надпочечников: 17-гидроксипрогестерон — 0,2 нмоль/л (норма 0,1–2,7 нмоль/л); кортизол — 768 нмоль/л (норма 140–600 нмоль/л).
Хлориды пота: 25,0; 21,5; 29,8 мэкв/л (норма ниже 40 мэкв/л).
Обследование на TORCH -комплекс: результат отрицательный.
Иммунограмма: лейк. — 8,9 х 10 9 /л, лимф. — 61 %, CD 3 — 70 %, CD22 — 24 %, CD4 — 43 %, CD8 — 27 %, CD25 — 19 %, IgА — 0,25 г/л, IgМ — 0,44 г/л, I gG — 5,4 г/л.
Результаты медико-генетического консультирования
Высокоэффективная жидкостная хроматография аминокислот крови: диагностически значимой патологии не выявлено.
ТСХ углеводов крови: нормограмма. ТСХ углеводов мочи: нормограмма.
Кариотип: 46, XY, 1 % хромосомной нестабильности (C- и G-окрашивание).
Содержание лактата в крови: 2,21 ммоль/л (норма 0,2–2,2 ммоль/л).
Содержание в моче мочевины, мочевой кислоты, КФК, ЛДГ: в пределах нормы.
Рентгенография органов грудной клетки: легочные поля вздуты, легочный рисунок усилен, обогащен на всем протяжении за счет гемодинамических нарушений. Реберно-диафрагмальные синусы свободны. Контуры купола диафрагмы ровные, четкие. Тень сердца не расширена. КТИ — 55 %.
Ультразвуковое исследование внутренних органов: печень увеличена на 4 см , эхогенность паренхимы повышена, структура однородна; поджелудочная железа не увеличена, эхогенность паренхимы повышена, структура однородна; добавочная селезенка 13 ´ 15 мм; врожденный порок сердца — коарктация аорты (зона сужения 3 мм на протяжении 4–5 мм), фракция выброса левого желудочка сердца — 66 %.
Невролог: у ребенка большой родничок на уровне костей черепа, не напряжен, зрачки D = S, реакция на свет вялая, непостоянный сходящийся страбизм, асимметрия лицевой мускулатуры, выражены стигмы дизэмбриогенеза лицевого черепа, мышечный тонус выраженно дистоничен, двигательная активность снижена, на осмотр реагирует хаотичным возбуждением, гримасой плача. Сухожильные рефлексы вызываются, торпидны. Патологические и менингеальные симптомы отсутствуют. Взгляд не фиксирует. Таким образом, у ребенка имеют место синдром двигательных нарушений, задержка психомоторного развития тяжелой степени.
Эндокринолог: данных в пользу наличия у ребенка адреногенитального синдрома, врожденного гипотиреоза не выявлено.
Хирург: правосторонняя паховая грыжа, неущемленная.
Иммунолог: убедительных данных в пользу наличия у ребенка иммунодефицита не выявлено.
Кардиолог: врожденный порок сердца — коарктация аорты, СН0.
Генетик: в фенотипе ребенка обращают на себя внимание выраженные черепно-лицевые дисморфии, гротескность черт лица (макростомия, гиперплазия альвеолярного отростка, готическое небо), широкий корень носа с вывернутыми ноздрями, длинный фильтр, низко расположенные увеличенные ушные раковины, расширенная венозная сеть на коже груди, живота, конечностей, головы, диспропорциональное телосложение, увеличенный живот, умеренная гепатомегалия. Рекомендуется проводить дифференциальную диагностику с синдромом Швахмана — Даймонда и митохондриальными болезнями.
За время наблюдения и лечения существенного улучшения в состоянии ребенка не отмечено. Ребенок перестал есть самостоятельно, вскармливался через зонд. Проводилась коррекция питания, менялись смеси, в том числе с высокой степенью гидролиза белка. В состав терапии вводились ферментные препараты, добавлялись продукты с высоким содержанием белка (творог). Однако положительного эффекта не наблюдалось. У ребенка прогрессировала гипотрофия, развивались явления кахексии, что свидетельствовало о наличии грубых метаболических нарушений. Учитывая прогрессирование гипотрофии и появление симптомов кахексии, трудности энтерального вскармливания, ребенку проводилось парентеральное питание в режиме гипералиментации растворами аминокислот, жировыми растворами, а также 12,5% раствором глюкозы. Указанная концентрация раствора глюкозы явилась максимально допустимой при круглосуточном введении, не вызывала глюкозурию при относительно невысоком уровне гликемии. Кроме парентерального питания проводилась коррекция водно-метаболических и электролитных нарушений. В дальнейшем, ввиду отсутствия положительной динамики, мать от продолжения лечения отказалась. При выписке ребенок получал энтерально через зонд высокоадаптированную молочную смесь.
Анализ результатов клинического наблюдения, лабораторно-инструментальных исследований и проведение дифференциальной диагностики с гипотрофией алиментарного генеза, адреногенитальным синдромом, врожденным гипотиреозом, хромосомными болезнями, муковисцидозом, первичным иммунодефицитом, TORCH-инфекциями, синдромом мальабсорбции, синдромом Швахмана — Даймонда, митохондриальными болезнями подтвердили наличие у ребенка врожденного порока сердца (коарктации аорты), грубых неврологических расстройств (задержки темпов психомоторного развития), правосторонней паховой грыжи, прогрессирующей гипотрофии, гипогликемии, что в совокупности с особенностями фенотипа ребенка позволило диагностировать у больного лепречаунизм.
Из катамнеза известно, что ребенок умер 24.04.2009 г. в возрасте 8 месяцев. От проведения аутопсии родители отказались.
Следует отметить, что литературные данные о лепречаунизме крайне немногочисленны. Впервые лепречаунизм описан в 1948 г . W. Donohue. Популяционная частота неизвестна, соотношение полов М1 : Ж3.
Название заболевания происходит от ирландского leprechaun — гном, так как больные имеют типичный внешний вид: гротескное лицо, гипертелоризм, выпуклые глаза, плоская переносица, большой рот с толстыми губами, расширенный кончик носа и большие ноздри; крупные, низко расположенные уши; микроцефалия (в некоторых случаях); арковидное небо; гирсутизм. Важный диагностический признак — увеличение молочных желез, клитора, половых губ у девочек. Возможны пупочные и паховые грыжи, расхождение прямых мышц живота, необычно большие кисти и стопы, гипотония, задержка окостенения. Дети с лепречаунизмом рождаются недоношенными, с внутриутробной гипотрофией. В связи с трудностями кормления больные крайне истощены, что приводит к появлению дополнительных кожных складок, особенно выраженных на конечностях. Характерна задержка психомоторного развития. Смерть наступает на первом году жизни.
У больных лепречаунизмом отмечаются гиперинсулинемия, гипогликемия, аминацидурия, накопление гликогена и железа в клетках печени; на аутопсии — гиперплазия и фолликулярные кисты яичников, гиперплазия островкового аппарата поджелудочной железы, гипертрофия и кальциноз почек. Нарушено связывание инсулина с рецепторами клетки, что обусловлено генетическими нарушениями, связанными с периферическим действием инсулина, являющимися следствием мутаций гена рецептора инсулина. Тип наследования — аутосомно-рецессивный. Обнаружены мутации в гене рецептора инсулина, локализованном на 19р13.3-р13.2 [1–3].
Описаны и другие формы лепречаунизма, такие как наследственная болезнь женщин, обусловленная нарушениями развития и дисфункцией эндокринной системы; характеризуется гипотрофией, малыми размерами лица, гирсутизмом, увеличением молочных желез, клитора и малых половых губ, гиперплазией яичников и островкового аппарата поджелудочной железы, накоплением гликогена и железа в печени, кальцинозом почек; наследуется по аутосомно-рецессивному типу, ограниченному полом [4].
Кроме этого, в современной классификации сахарного диабета лепречаунизм описан как другой тип сахарного диабета, связанный с генетическим дефектом в действии инсулина [5].
Анализ материалов данного клинического наблюдения позволяет считать, что диагностика лепречаунизма представляет собой значительные трудности ввиду крайне низкой частоты встречаемости данной патологии, многие аспекты этиологии и патогенеза которой требуют дальнейшего изучения. Описанный случай поможет ознакомить врачей с особенностями клинических проявлений лепречаунизма у ребенка раннего возраста.
1. Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование: Справочник. — Л.: Медицина, 1987. — 320 с.
2. Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование: Атлас-справочник. — 3-е изд., перераб. и доп. — М.: Т-во научных изданий КМК: Авторская академия, 2007. — 448 с.
3. Wikipedia, the free encyclopedia. Donohue syndrome. http://en.wikipedia.org/wiki/Donohue_syndrome
4. Энциклопедический словарь медицинских терминов. http://www.medicalipedia.com/1/203/206608.html
5. Балаболкин М.И., Клебанова Е.М., Креминская В.М. Диагностика и классификация сахарного диабета // Сахарный диабет. — 1999. — Т. 4, № 3. http://www.diabet.ru/Sdiabet/1999-03/3.htm